Il prione cos’è? Caratteristiche generali
Le proteine prioniche o prioni (PrPC) sono proteine costitutivamente espresse a livello del sistema nervoso centrale, in particolar modo sulla superficie delle membrane cellulari di neuroni, cellule della microglia, astrociti, oligodendrociti e sulle cellule muscolari scheletriche. Le proteine dei prioni vengono codificate dal gene PRNP che mappa sul braccio corto del cromosoma 20 ed è costituita da 254 aminoacidi. Una volta sintetizzati a livello del reticolo endoplasmatico, i prioni, essendo proteine secretorie, prima che vengano espresse sulla superficie della membrana cellulare subiscono delle modifiche post-traduzionali (taglio del peptide segnale e glicosilazione). Le mutazioni puntiformi come le sostituzioni o le inserzioni sono alla base del misfolding e determinano un cambiamento della struttura secondaria dei prioni generando così la forma patologica (PrPSC), agente eziologico di un gruppo di patologie neurodegenerative ad esito fatale note come encefalopatie spongiformi trasmissibili (EST).
Struttura dei prioni
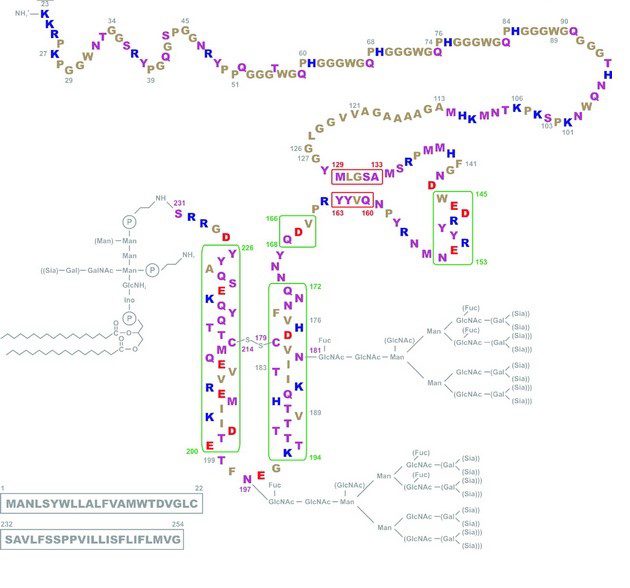
La PrPC (fig.1) è localizzata a livello di microdomini noti come zattere lipidiche sulla superficie esterna della membrana plasmatica tramite un’ancora di glicosil-fosfatidil-inositolo (GPI). Quest’ultima è legata al residuo di serina 231 della proteina. All’estremità N-terminale è presente un peptide segnale (aa 1-22) e quattro ripetizioni identiche di un octapeptide. La presenza di residui di istidina (H) rende questa regione ad alta affinità nel legare ioni rame e per tal motivo si pensa che il prione possa avere un ruolo nella regolazione della concentrazione di questi ioni in corrispondenza delle fessure sinaptiche. I residui di asparagina 181 e 197 sono siti di glicosilazione, per cui la proteina può trovarsi nelle isoforme non glicosilata, monoglicosilata e diglicosilata. Il dominio globulare di PrPC (aa 125-228) comprende tre regioni α-elica e due a foglietto β antiparallelo. Sono le interazioni non covalenti come i legami idrogeno o le interazioni idrofobiche di questi residui ad essere le responsabili durante il folding della struttura terziaria della proteina. In questo dominio inoltre è presente un ponte disolfuro tra due residui di cisteina in posizione 179 e 214.
Proteina prionica patologica PrPSC
Questa proteina si forma in seguito al misfolding dei prioni e di conseguenza anche la sua struttura secondaria risulta modificata, infatti se nella proteina prionica normale il contenuto di α-eliche è nettamente superiore (42%) a quello dei foglietti β (3%), nella PrPSC si osserva l’opposto, con un contenuto di foglietti β intorno al 40% e una piccola percentuale in α-elica (fig.2).
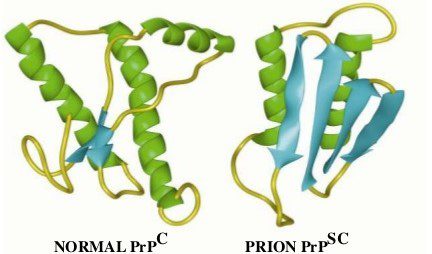
Questa diversa conformazione determina nuove proprietà chimico-fisiche alla proteina prionica patologica quali: insolubilità, tendenza all’aggregazione (formazione di fibrille) e resistenza alla proteolisi. Quando la proteina prionica patologica entra in contatto con quella fisiologica, induce quest’ultima ad adottare la propria conformazione. Ciò indica che la formazione di PrPSC è autocatalitica, ovvero la PrPSC formatasi inizialmente stimola la produzione di ulteriore PrPSC a partire da PrPC. Questo tipo di meccanismo con il quale la proteina “si replica” è stato avvalorato dal fatto che topi knockout per il gene della proteina prionica risultano resistenti alle encefalopatie spongiformi trasmissibili.
Encefalopatie Spongiformi Trasmissibili (EST)
Come si è già detto le EST sono un gruppo di patologie neurodegenerative, possono colpire l’uomo e altri mammiferi. Possono presentarsi in forma sporadica, genetica o familiare e acquisita. Negli animali ad oggi si conoscono: lo scrapie delle pecore e capre, la malattia cronica debilitante (CWD) dei cervidi, l’encefalopatia spongiforme bovina (BSE), l’encefalopatia trasmissibile del visone (TME) e l’encefalopatia spongiforme felina (FSE) del gatto. Nell’uomo sono note le forme sporadica, familiare, iatrogena e una variante della malattia di Creutzfeldt-Jakob (CJD), la sindrome di Gerstmann-Sträussler-Scheinker (GSS), l’insonnia fatale nelle forme sporadica (sFI) e familiare (FFI) ed il kuru.
Scrapie
Lo scrapie delle pecore e capre si manifesta clinicamente con disturbi dell’equilibrio, tremori ed intenso prurito (il nome della malattia deriva dal verbo “to scrape” che significa grattare/raschiare) che induce gli animali a provocarsi ferite anche profonde strofinandosi contro qualunque superficie. Colpisce animali di età compresa tra i 2 e i 4 anni ed il periodo d’incubazione è compreso tra i 2 e 5 anni. Negli animali colpiti da scrapie, PrPSC è stato isolato nell’encefalo, tratto intestinale e ghiandole salivari.
Encefalopatia spongiforme bovina (BSE)
L’encefalopatia spongiforme bovina (BSE) si manifesta dopo un periodo di incubazione compreso tra 4 e 6 anni in bovini di età superiore ai 4 anni. I segni clinici sono caratterizzati da tremori muscolari, disturbi del comportamento e iperestesia a stimoli tattili (fig.3). Viene anche chiamata morbo della mucca pazza.
Malattia di Creutzfeldt-Jakob (CJD) sporadica
La Malattia di Creutzfeldt-Jakob (CJD) sporadica presenta un quadro clinico caratterizzato da disturbi cognitivi e del comportamento. I segni neurologici più frequenti sono di natura cerebellare quali, disturbi dell’equilibrio e visivi (nistagmo e diplopia). I soggetti affetti da CJD sporadica hanno una sopravvivenza media di 6 mesi dall’inizio dei primi sintomi. Il principale polimorfismo responsabile della diversa suscettibilità alla CJD sporadica nella popolazione è quello relativo al codone 129, che può codificare per metionina (M) o valina (V). Studi epidemiologici hanno dimostrato che questa patologia tende a manifestarsi maggiormente negli individui omozigoti MM e VV. Nella Malattia di Creutzfeldt-Jakob familiare la causa è attribuibile a mutazioni puntiformi del gene PRNP (sostituzioni). Alcuni esempi di mutazioni più frequenti osservate in casi italiani sono: E200K, V203I, R208H e V210I. La nuova variante della malattia di Creutzfeld-Jakob (vCJD) è correlata al consumo di carne bovina proveniente da animali affetti da BSE (fig.4). Fu diagnosticata per la prima volta nel 1996 in individui con età media intorno ai 30 anni. Dal 1996 al 2004 sono stati segnalati circa 152 casi, molti dei quali nel Regno Unito. La sintomatologia è simile alla forma sporadica.
Sindrome di Gerstmann-Sträussler-Scheinker (GSS)
La sindrome di Gerstmann-Sträussler-Scheinker (GSS) è una malattia autosomica dominante e generalmente si manifesta negli individui con età superiore ai 40 anni. Caratterizzata da disfunzione cerebellare e demenza. La causa della patologia è dovuta ad alcune mutazioni del gene PRNP e riguardano: P102L, P105L e A117V.
Insonnia familiare fatale (FFI)
Insonnia familiare fatale (FFI). Malattia autosomica dominante che colpisce alcune aree della corteccia cerebrale ed i nuclei talamici, provocando gravi disturbi del ritmo sonno-veglia nel soggetto affetto. Il corso della malattia in genere è di un anno provocando inevitabilmente la morte del paziente. La causa della patologia è dovuta anche in questo caso a mutazioni puntiformi nel gene PRNP.
Diagnosi
La diagnosi di certezza di tutte le malattie da prioni si basa sull’esame istologico del cervello e sull’identificazione immunochimica di PrPSC. Per la diagnosi in vita della CJD sporadica si ricorre a esami strumentali e criteri clinici come l’EEG e l’identificazione della proteina 14-3-3 (isoforme). Anomalie dell’EEG caratteristiche di tale patologia sono i complessi periodici trifasici punta-onda (1-2 cicli al secondo) sincroni e bilaterali, anche se non sono costanti durante l’intera fase clinica. L’SDS-PAGE e l’immunoblot sono le tecniche di routine impiegate per l’identificazione della proteina 14-3-3. Questa è un marker generico di distruzione tissutale del cervello e la si ritrova nel liquido cerebrospinale. Per la diagnosi delle forme genetiche è molto importante la sequenza del gene PRNP.
Si ringrazia Diego Piacentini per il gentile contributo
Fonti
- M.D. Geschwind, 2015, Prionic Diseases.
- A.S. Das & W. Zou, 2016, Prions: beyond a single protein.
- S. Capellari, R. Strammiello, D. Saverioni et all., 2011, Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: insights into phenotypic variability and disease pathogenesis.
- G. Antonelli & M. Clementi, Principi di virologia medica, 2008.
- http://www.stanford.edu
- http://www.who.int
- www.scienzagiovane.unibo.it
- https://www.microbiologiaitalia.it