Descrizione della malattia
La sindrome di Marfan è una malattia rara ereditaria che colpisce il tessuto connettivo causando alterazioni cardiache, dei vasi sanguigni, polmonari, oculari, ossee e del sistema nervoso centrale. Questa malattia prende il nome dal pediatra francese Antoine Marfan, il quale la descrisse per la prima volta nel 1896 in una bambina di 5 anni. Si è dovuto attendere fino al 1991 affinché il dottor Francesco Ramirez identificasse il gene alterato coinvolto nella malattia.
La sindrome di Marfan è una malattia genetica autosomica dominante provocata da un’alterazione del gene FBN1, presente sul cromosoma 15, che codifica per la fibrillina-1, una glicoproteina essenziale del tessuto connettivo che rappresenta il supporto strutturale delle microfibrille. Le microfibrille sono costituite da fibrillina e sono presenti nella matrice extracellulare dove formano un intreccio per la deposizione dell’elastina nelle fibre elastiche. Esse sono presenti in tutto l’organismo, tuttavia si trovano soprattutto a livello del sistema cardiovascolare, scheletrico e oculare, i principali bersagli della malattia (Fig. 1).
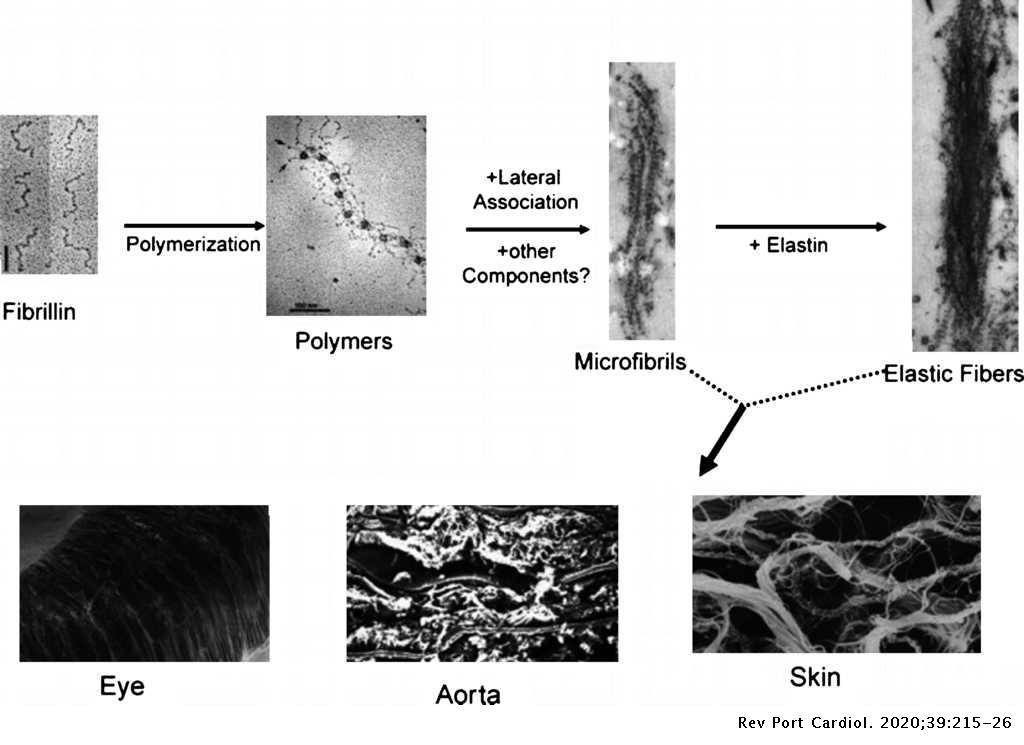
Alcune forme cliniche di confine sono dovute alle mutazioni del gene TGFBR2, localizzato sul cromosoma 3, che codifica per un recettore del fattore di crescita trasformante beta (TGF-beta).
Epidemiologia
La prevalenza della malattia è stimata 1:5000 senza differenza tra i sessi. Molto probabilmente l’incidenza della malattia è sottostimata per via delle frequenti forme atipiche che, non manifestandosi con un fenotipo peculiare, possono non essere diagnosticate. Circa tre quarti dei pazienti affetti da sindrome di Marfan ha almeno un genitore affetto a sua volta con trasmissione autosomica dominante. Un quarto dei pazienti non ha familiarità con la malattia; in questo caso si tratta di una mutazione genetica de novo che si è verificata negli spermatozoi paterni.
Una persona affetta ha una probabilità del 50% di trasmettere la mutazione patogenetica. La diagnosi genetica prenatale è possibile nelle famiglie nelle quali è stata identificata la mutazione responsabile della malattia.
Caratteristiche cliniche
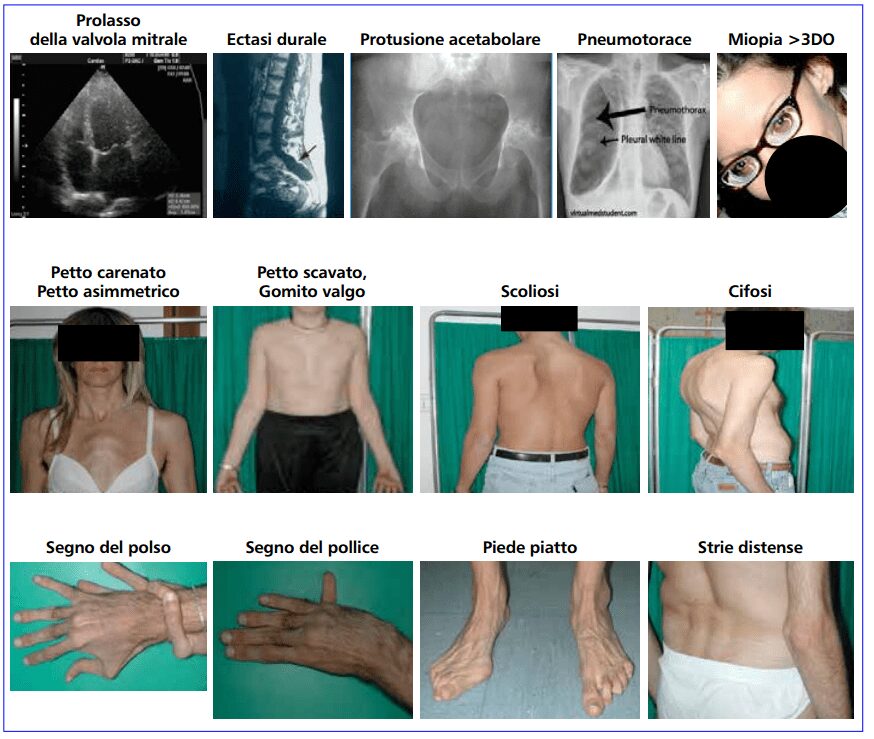
I pazienti affetti da sindrome di Marfan presentano delle caratteristiche peculiari in molte parti del corpo anche se è raro che una persona le manifesti tutte (Fig. 2). Alcune di esse sono semplici da riconoscere visivamente, altre, invece, possono essere individuate solo tramite esami.
Sistema cardiovascolare
A livello del sistema cardiovascolare i reperti principali riscontrati nei pazienti sono l’aneurisma aortico e il prolasso valvolare (Figura 3).
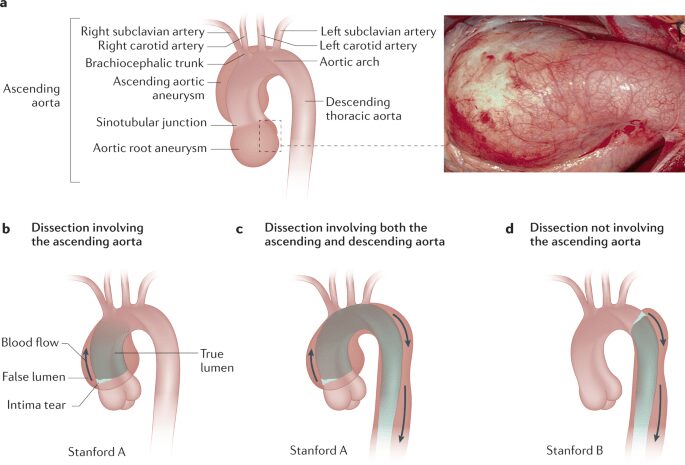
Le complicanze più gravi sono provocate da alterazioni nella radice aortica e nell’aorta ascendente. L’aorta viene colpita a livello della tonaca media, principalmente in zone soggette a maggiore stress emodinamico; essa si dilata progressivamente o si disseca in acuto, a partire dai seni coronarici. È molto comune riscontrare anomalie a carico della valvola mitrale e della valvola tricuspide. Queste affezioni a livello valvolare sono dovute o al prolasso dei lembi oppure alla rottura delle corde tendinee. In questi casi sono spesso presenti rumori caratteristici e rigurgiti che possono progredire portando nei casi più gravi alla deiscenza della valvola. In alcune circostanze può risultare essenziale un intervento chirurgico e la sostituzione della valvola con una meccanica oppure una biologica, con un tasso di sopravvivenza molto positivo.
Nei pazienti affetti dalla malattia si possono verificare anche aritmie ventricolari importanti che possono aumentare il rischio di morte cardiaca improvvisa e arresto cardiaco.
Apparato muscolo-scheletrico
Per quel che riguarda le caratteristiche della malattia a livello delle ossa e delle articolazioni, la gravità varia notevolmente. I pazienti sono più alti della norma rispetto alla loro età e ai loro familiari, spesso sviluppano segni di magrezza eccessiva; l’apertura delle braccia supera l’altezza. L’aracnodattilia (“dita a ragno”) è molto comune e notevole, le dita risultano affusolate e sproporzionalmente allungate rispetto al palmo della mano o alla pianta del piede. Il “segno del pollice” (la falange distale del pollice supera il margine del pugno chiuso) e il “segno del polso” (le falangi distali di pollice e mignolo si sovrappongono quando poste a cingere il polso) sono caratteristiche distintive della malattia. Le malformazioni dello sterno, il torace carenato (spostamento all’esterno), il pectus excavatum (spostamento all’interno) sono frequenti. Molto comuni sono anche l’ipersensibilità delle articolazioni, il genu recurvatum (curvatura all’indietro delle gambe a livello delle ginocchia), i piedi piatti, la cifoscoliosi e le ernie inguinali e diaframmatiche.
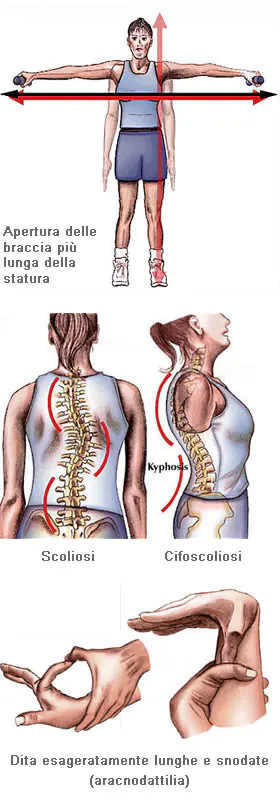
Altri apparati e organi
Le manifestazioni a livello oculare sono essenzialmente due, il dislocamento del cristallino (ectopia lentis) e la sferofachia che porta al distacco della retina. Si possono riscontrare anche glaucoma a esordio precoce o cataratta. A livello polmonare possono insorgere pneumopatia cistica e pneumotorace spontaneo ricorrente. Nel sistema nervoso centrale l’ectasia durale (rigonfiamento del sacco durale che circonda il midollo spinale) è un reperto che compare frequentemente in questa malattia e colpisce in genere la colonna lombo-sacrale. Può causare cefalea, dolore lombare o deficit neurologici che si manifestano con debolezza intestinale o vescicale.
I soggeti affetti dalla sindrome sono più proni a sviluppare endocardite infettiva, per questo motivo in caso di interventi o sospette infezioni e sempre meglio una profilassi antibiotica preventiva.
Diagnosi e test
Considerate le oltre 200 mutazioni possibili, l’impiego di marker genetici risulta pressoché improponibile a finalità diagnostiche. La diagnosi della malattia si basa sui segni clinici e sulla storia familiare anche se è difficile formularla, in quanto i quadri clinici sono molto variabili.
Per favorire una corretta diagnosi della malattia, sono stati proposti dei criteri internazionali (criteri di Ghent, formulati nel 1996 e aggiornati nel 2010) fondati su una valutazione completa basata in gran parte su una combinazione di manifestazioni cliniche maggiori e minori in vari sistemi d’organo e sulla storia familiare.
I criteri di Ghent prevedono un sistema a punteggio (Figura 5).
Vengono considerati criteri maggiori: la dilatazione, l’aneurisma o la dissecazione dell’aorta toracica, la sublussazione o lussazione del cristallino, caratteristiche sistemiche che raggiungano un punteggio=/> 7 e presenza di un parente di primo grado affetto dalla malattia.

Unitamente a questo sistema diagnostico si effettuano anche test ed esami clinici, come:
- Ecocardiografia/RM per misurare la radice aortica ed individuare il prolasso valvolare.
- Angiorisonanza magnetica e angiografia con mezzo di contrasto per rendere evidenti le strutture interne dell’aorta.
- Esame con la lampada a fessura per riscontrare anomalie del cristallino, come la lussazione, e misura della pressione oculare per identificare l’eventuale presenza di glaucoma.
- RX del sistema scheletrico di mano, colonna vertebrale, bacino, torace, piede e cranio alla ricerca di anomalie caratteristiche.
- RM del rachide lombosacrale (ectasia durale).
La sindrome di Marfan può incorrere in diagnosi differenziale con altre malattie genetiche e connettivopatie.
Terapie
Il trattamento della sindrome di Marfan è focalizzato sulla prevenzione della complicanze.
Le principali cause di morte legate a questa sindrome sono da ricondursi alle manifestazioni che si verificano a carico dell’apparato cardiocircolatorio, in particolare la dissecazione aortica e gli aneurismi. Al fine di evitare che l’aorta si indebolisca e si allarghi, è importante ridurre la pressione sanguigna. Per ottenere questo risultato, i farmaci di prima scelta che i pazienti devono assumere sono i betabloccanti, che rallentano la frequenza cardiaca e ritardano l’onda aortica, riducono la contrattilità miocardica e la pressione differenziale, e rallentano la progressione della dilatazione della radice aortica e il rischio di dissezione.
Ultimamente la pratica clinica ha evidenziato che si ottiene una maggior beneficio con i sartani (tendono a rallentare in modo significativo la dilatazione dell’aorta ascendente) e con gli ACE inibitori (possono avere un ruolo costante nella riduzione dell’espansione ritardata nell’arco e nell’aorta addominale).
La chirurgia preventiva è consigliata quando il diametro aortico supera i 5 cm. Nelle donne in gravidanza il rischio di complicanze aortiche è moto elevato, quindi la riparazione aortica elettiva deve essere valutata prima del concepimento.
Le problematiche a livello dell’apparato scheletrico sono gestite con l’utilizzo di corsetti correttivi. È consigliato l’intervento chirurgico nei pazienti con curve di 40-50°.
Prognosi
Negli anni ’70 la mortalità per gli uomini si aggirava intorno ai 40 anni, per le donne era lievemente più alta. Con gli anni, il miglioramento dei metodi di diagnosi e di screening, un adeguato stile di vita, i trattamenti farmacologici e chirurgici hanno migliorato la qualità e la sopravvivenza e per gli uomini l’aspettativa di vita si è alzata a 62 anni e nelle donne a 73 anni.
Fonti
- Milewicz et al, Marfan syndrome, Nature Reviews Disease Primers volume 7, Article number: 64 (2021)
- Manuale MSD
- Loey’s et al. The revised Ghent nosology for the Marfan syndrome, J Med Genet, 2010 Jul;47(7):476-85.
- https://www.orpha.nethttps://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=it&Expert=558
- https://www.grupposandonato.it/news/2022/gennaio/sindrome-di-marfan-cose-sintomi
- https://www.ospedalebambinogesu.it/sindrome-di-marfan-80377/
Crediti immagini
- Figura 1- Ramirez F, Sakai LY, Dietz HC, et al., Physiol Genomics, 19(2): 151‐4, 2004
- Figura 2 – Loey’s et al. The revised Ghent nosology for the Marfan syndrome, J Med Genet, 2010 Jul;47(7):476-85.
- Figura 3 – Milewicz et al, Marfan syndrome, Nature Reviews Disease Primers volume 7, Article number: 64 (2021)
- Figura 4 – mypersonaltrainer
- Figura 5 – Loey’s et al. The revised Ghent nosology for the Marfan syndrome, J Med Genet, 2010 Jul;47(7):476-85.