Nel mondo della patologia craniofacciale, l’acrocefalosindattilia emerge come una condizione rara ma significativa. Questo articolo fornisce un’esplorazione approfondita delle caratteristiche, delle cause, dei sintomi, della diagnosi, della prevenzione e delle opzioni di trattamento legate all’acrocefalosindattilia. Attraverso questa panoramica, speriamo di offrire una comprensione completa di questa patologia, così come delle opzioni disponibili per affrontarla.
Panoramica sull’Acrocefalosindattilia
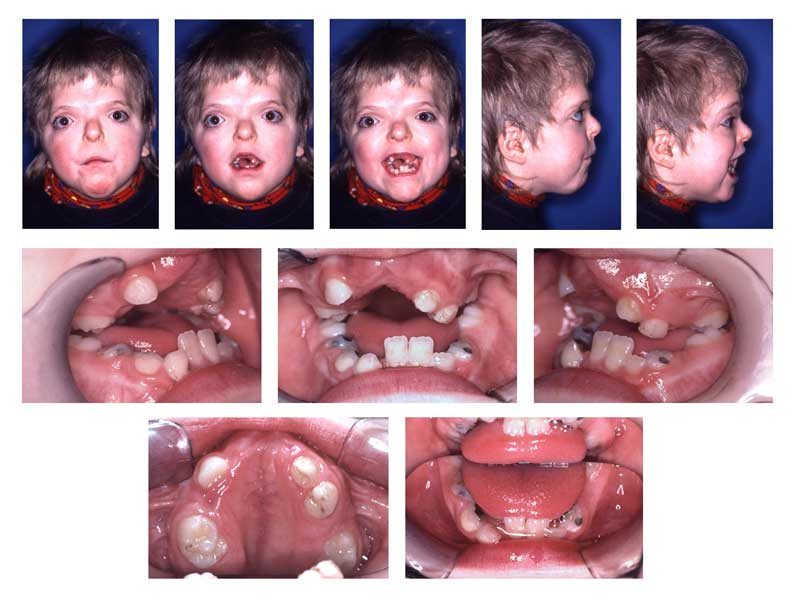
L’acrocefalosindattilia, noto anche come sindrome di Apert, è una malattia genetica rara che colpisce lo sviluppo craniofacciale e scheletrico. È caratterizzato dalla fusione prematura delle ossa craniche e da anormalità nei tessuti molli del viso e delle mani. La sindrome di Apert è risultato dell’autosomico dominante, il che significa che un solo genitore portatore del gene mutato può trasmettere la condizione al proprio figlio.
Cause
L’acrocefalosindattilia è causata da mutazioni nel gene FGFR2 che regola la crescita e lo sviluppo delle ossa e dei tessuti connettivi. Queste mutazioni portano a un’attivazione inappropriata della via di segnalazione del recettore del fattore di crescita dei fibroblasti (FGFR), che a sua volta influisce sulla normale crescita ossea e tessutale. La maggior parte dei casi di acrocefalosindattilia non è ereditata da genitori affetti, ma è il risultato di nuove mutazioni spontanee.
Sintomi dell’Acrocefalosindattilia
I sintomi dell’acrocefalosindattilia sono diversificati e interessano principalmente il cranio, il viso e le mani. Tra i sintomi più comuni ci sono:
- Craniostenosi: Fusione prematura delle suture craniche, causando una testa anormalmente formata.
- Ipertelorismo: Un’ampia distanza tra gli occhi.
- Midface retrusion: Il mezzo del viso appare spinto all’indietro.
- Sindattilia: Fusione delle dita delle mani e, talvolta, dei piedi.
- Alterazioni dentali: Denti sovrapponibili e altre anomalie dentali.
- Orecchie alte e deformate: Posizionamento anomalo delle orecchie.
Diagnosi dell’Acrocefalosindattilia
La diagnosi dell’acrocefalosindattilia coinvolge una valutazione clinica approfondita, inclusa l’analisi dei sintomi fisici caratteristici. Le immagini diagnostiche, come la tomografia computerizzata (TC) e la risonanza magnetica (RM), sono essenziali per valutare la struttura cranica e rilevare anomalie interne. L’identificazione delle mutazioni del gene FGFR2 conferma la diagnosi.
Prevenzione dell’Acrocefalosindattilia
Poiché la sindrome di Apert è il risultato di mutazioni genetiche spontanee, la prevenzione primaria si concentra sull’educazione e sulla consulenza genetica. Le famiglie con una storia di acrocefalosindattilia possono essere informate sui rischi di ricorrenza e sottoporsi a test genetici prenatali durante la gravidanza.
Trattamento dell’Acrocefalosindattilia
Il trattamento dell’acrocefalosindattilia richiede un approccio multidisciplinare. Interventi chirurgici precoci sono spesso necessari per correggere le anomalie craniche e facciali, nonché per separare le dita delle mani fuse. La terapia dell’udito e il trattamento ortodontico possono essere necessari nel lungo periodo. Il supporto psicologico è fondamentale per i pazienti e le loro famiglie, affrontando le sfide estetiche e funzionali associate alla condizione.
Conclusione
In sintesi, l’acrocefalosindattilia è una condizione patologica complessa che richiede una gestione interdisciplinare e tempestiva. La comprensione delle cause, dei sintomi e delle opzioni di trattamento è fondamentale per garantire una migliore qualità di vita ai pazienti colpiti. Attraverso l’educazione, la ricerca e il sostegno, possiamo affrontare le sfide presentate da questa sindrome e lavorare verso una migliore prognosi.
Fonte: National Organization for Rare Disorders (NORD)