Scopri la sindrome di Mowat-Wilson, una malattia genetica rara con caratteristiche uniche e implicazioni per la salute.
Indice
- Introduzione alla Sindrome di Mowat-Wilson (malattia genetica)
- Cause genetiche della sindrome di Mowat-Wilson
- Caratteristiche cliniche e fenotipo della malattia genetica
- Anomalie associate alla sindrome di Mowat-Wilson
- Diagnosi della sindrome di Mowat-Wilson
- Gestione e trattamento della condizione genetica
- Prospettive di ricerca sulla malattia genetica rara
- Conclusioni su sindrome di Mowat-Wilson
- Domande Frequenti su sindrome di Mowat-Wilson
- Leggi anche:
- Fonti
- Crediti fotografici
- Segui Microbiologia Italia
Questo articolo esplora in modo completo la sindrome di Mowat-Wilson, una condizione genetica rara caratterizzata da disabilità intellettiva, tratti facciali distintivi e malformazioni multiple. Tratteremo cause genetiche, sintomi clinici, diagnosi, gestione e prospettive future. Può essere utile per famiglie, medici, genetisti e caregiver interessati alla malattia genetica rara, offrendo strumenti per riconoscere precocemente la condizione e migliorare la qualità della vita dei pazienti.
Introduzione alla Sindrome di Mowat-Wilson (malattia genetica)
La sindrome di Mowat-Wilson (MWS) rappresenta una malattia genetica complessa e poco frequente, causata da alterazioni nel gene ZEB2. Descritta per la prima volta nel 1998, questa patologia si manifesta con un fenotipo cranio-facciale tipico, ritardo nello sviluppo e anomalie congenite variabili.
La sindrome di Mowat-Wilson colpisce principalmente il sistema nervoso centrale, l’apparato gastrointestinale e altri organi, rendendo essenziale una diagnosi multidisciplinare. Comprendere questa condizione genetica rara aiuta a orientare interventi tempestivi e supporto familiare.
Nel corso dell’articolo analizzeremo i meccanismi molecolari, i segni clinici e le strategie di cura, con enfasi su variazioni semantiche come disordine genetico di Mowat-Wilson, patologia ZEB2-related e sindrome con haploinsufficienza ZEB2.
Cause genetiche della sindrome di Mowat-Wilson
La sindrome di Mowat-Wilson deriva da mutazioni eterozigoti o delezioni nel gene ZEB2 (Zinc finger E-box binding homeobox 2), localizzato sul cromosoma 2q22.3. Questo gene codifica un fattore di trascrizione essenziale per lo sviluppo embrionale di tessuti neurali, cardiaci e intestinali.
Nella maggior parte dei casi, si tratta di mutazioni de novo che provocano haploinsufficienza di ZEB2, ovvero la perdita di funzione di una copia del gene. Oltre 100 varianti patogene sono state identificate, incluse delezioni parziali o totali del gene e mutazioni troncanti.
La sindrome di Mowat-Wilson segue un pattern di ereditarietà autosomica dominante, ma i casi familiari sono rarissimi poiché le mutazioni sorgono spontaneamente. In rari pazienti con fenotipo atipico, varianti missenso possono interessare domini funzionali specifici della proteina.
Studi molecolari confermano che l’aploinsufficienza di ZEB2 altera la migrazione delle cellule della cresta neurale, spiegando l’associazione frequente con la malattia di Hirschsprung e altre malformazioni.
Caratteristiche cliniche e fenotipo della malattia genetica
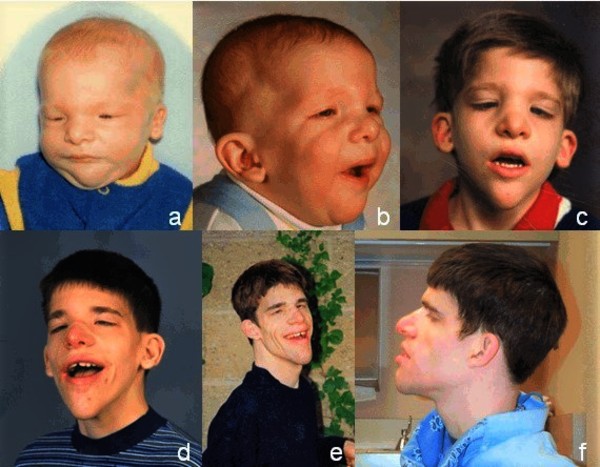
Il fenotipo della sindrome di Mowat-Wilson evolve con l’età e include tratti facciali distintivi che diventano più evidenti nel tempo. Tra questi spiccano fronte alta, sopracciglia folte e divergenti medialmente, ipertelorismo, occhi infossati, naso a sella con punta arrotondata, columella prominente, bocca aperta con labbro superiore a M e mento appuntito e prominente. Le orecchie appaiono sollevate con una depressione centrale, spesso definite “a coppa”.
La sindrome di Mowat-Wilson si accompagna quasi sempre a disabilità intellettiva da moderata a grave, con ritardo psicomotorio e linguaggio assente o limitato a poche parole. L’epilessia rappresenta un segno cardine, presente in quasi tutti i pazienti oltre i 5 anni di età.
Altre manifestazioni comuni includono microcefalia, bassa statura, agenesia o ipoplasia del corpo calloso e anomalie oculari come macroftalmia o difetti della vista. I bambini mostrano spesso una soglia del dolore elevata e un comportamento allegro con frequenti risate.
Anomalie associate alla sindrome di Mowat-Wilson
Oltre ai tratti neurologici, la patologia genetica Mowat-Wilson coinvolge molteplici apparati. La malattia di Hirschsprung (HSCR) compare nel 40-50% dei casi, causando ostruzione intestinale congenita per assenza di cellule ganglionari nel colon.
Frequenti sono anche cardiopatie congenite (difetti settali, stenosi polmonare), anomalie genitourinarie (ipospadia nei maschi, malformazioni renali) e difetti muscolo-scheletrici. In alcuni pazienti si osservano asplenia o iposplenia, sebbene rare.
La sindrome di Mowat-Wilson può presentare malformazioni cerebrali strutturali visibili alla RMN, come agenesia del corpo calloso o alterazioni della sostanza bianca. Queste contribuiscono al quadro neurologico complesso.
La variabilità fenotipica è elevata: mentre i dismorfismi facciali e il ritardo dello sviluppo sono costanti, le malformazioni gravi variano da individuo a individuo.
Diagnosi della sindrome di Mowat-Wilson
La diagnosi di sindrome di Mowat-Wilson inizia con il riconoscimento clinico del fenotipo facciale caratteristico associato a disabilità intellettiva e convulsioni. L’esame obiettivo e la storia familiare (spesso negativa per casi ereditari) orientano verso questa malattia genetica.
La conferma avviene attraverso test molecolari sul gene ZEB2: sequenziamento diretto, MLPA per delezioni o array-CGH per riarrangiamenti cromosomici più ampi. Il gold standard è l’analisi genetica mirata che identifica mutazioni patogene nell’84% dei casi tipici.
La sindrome di Mowat-Wilson va differenziata da altre sindromi con ritardo mentale e dismorfismi, come quelle da mutazioni in altri geni della cresta neurale. La diagnosi precoce, possibile già in età infantile, permette un intervento multidisciplinare ottimale.
Gestione e trattamento della condizione genetica
Non esiste una cura causale per la sindrome di Mowat-Wilson, ma la gestione è sintomatica e multidisciplinare. Per l’epilessia si utilizzano antiepilettici standard, monitorando attentamente la risposta poiché le crisi possono essere refrattarie.
La malattia di Hirschsprung richiede intervento chirurgico di pull-through per rimuovere il segmento agangliare. Cardiopatie congenite e anomalie genitourinarie necessitano di correzioni chirurgiche tempestive.
La sindrome di Mowat-Wilson beneficia di riabilitazione motoria, logopedia e supporto cognitivo fin dai primi mesi di vita. Terapie occupazionali aiutano a sviluppare autonomie residue. Il follow-up include controlli cardiologici, gastroenterologici, neurologici e oculistici regolari.
Una soglia del dolore elevata richiede attenzione particolare per non sottovalutare traumi o patologie interne. Il supporto psicologico alle famiglie è fondamentale per affrontare la complessità della patologia ZEB2-related.
Prospettive di ricerca sulla malattia genetica rara
La ricerca sulla sindrome di Mowat-Wilson avanza grazie a modelli cellulari (iPSC derivati da pazienti) che riproducono difetti nella specifica neuronale e nella migrazione delle cellule della cresta neurale. Questi studi chiariscono il ruolo di ZEB2 nella regolazione trascrizionale durante lo sviluppo embrionale.
Terapie innovative, come approcci di editing genetico o potenziamento della funzione residua di ZEB2, rappresentano orizzonti futuri. Registri internazionali e studi genotipo-fenotipo aiutano a definire linee guida di cura personalizzate.
La sindrome di Mowat-Wilson evidenzia l’importanza della genetica medica nella medicina di precisione. La collaborazione tra centri di riferimento per malattie rare favorisce diagnosi più rapide e migliori outcomes a lungo termine.
Conclusioni su sindrome di Mowat-Wilson
In sintesi, la sindrome di Mowat-Wilson è una malattia genetica rara ma riconoscibile grazie al suo fenotipo facciale caratteristico, alla disabilità intellettiva e alle anomalie congenite multiple legate a mutazioni nel gene ZEB2.
Nonostante la gravità del quadro, una diagnosi precoce e una gestione multidisciplinare migliorano significativamente la qualità della vita dei pazienti e delle famiglie. La condizione genetica di Mowat-Wilson sottolinea il valore della ricerca traslazionale per trasformare la comprensione molecolare in interventi concreti.
La sindrome di Mowat-Wilson rimane una sfida, ma l’impegno scientifico e il supporto comunitario offrono speranza per un futuro migliore.
Domande Frequenti su sindrome di Mowat-Wilson
Chi colpisce principalmente la sindrome di Mowat-Wilson? La sindrome di Mowat-Wilson colpisce indifferentemente maschi e femmine, con casi sporadici in tutto il mondo. Consiglio: consulta un genetista pediatrico se sospetti tratti facciali tipici associati a ritardo dello sviluppo.
Cosa causa esattamente la sindrome di Mowat-Wilson? La causa è l’haploinsufficienza del gene ZEB2 sul cromosoma 2, dovuta a mutazioni o delezioni de novo. Consiglio: richiedi sempre il test molecolare ZEB2 per confermare la diagnosi clinica.
Quando si manifesta la sindrome di Mowat-Wilson? I segni compaiono spesso alla nascita o nei primi anni di vita, con epilessia che emerge tipicamente dopo i 5 anni. Consiglio: monitora lo sviluppo neurologico fin dai primi mesi per interventi precoci.
Come si diagnostica la sindrome di Mowat-Wilson? Attraverso valutazione clinica del fenotipo e conferma genetica con sequenziamento o MLPA del gene ZEB2. Consiglio: rivolgersi a centri specializzati in malattie rare per un approccio multidisciplinare accurato.
Dove trovare supporto per la sindrome di Mowat-Wilson? Associazioni di famiglie, centri di riferimento per malattie rare e siti come Orphanet offrono risorse e comunità. Consiglio: unisciti a gruppi di supporto per condividere esperienze e accedere a informazioni aggiornate.
Perché è importante conoscere la sindrome di Mowat-Wilson? La conoscenza permette diagnosi tempestiva, gestione ottimale delle complicanze e counseling genetico familiare. Consiglio: informa pediatri e specialisti sui tratti distintivi per evitare ritardi diagnostici.
Leggi anche:
Fonti
- https://pubmed.ncbi.nlm.nih.gov/34199024/ (Neurological Phenotype of Mowat-Wilson Syndrome)
- https://pubmed.ncbi.nlm.nih.gov/20301585/ (Classic Mowat-Wilson Syndrome)
- https://www.ncbi.nlm.nih.gov/books/NBK1412/ (GeneReviews – Classic Mowat-Wilson Syndrome)
Crediti fotografici
Immagine in evidenza – Link
{kind=link}
Segui Microbiologia Italia
Se ti è piaciuto questo contenuto e vuoi supportare Microbiologia Italia seguici anche su MSN e su Google News.