La progettazione e lo sviluppo dei prodotti farmaceutici richiedono tempi lunghi e costi elevati. Da qualche decennio, l’utilizzo di metodi bioinformatici, associato alle tecniche di genomica funzionale e agli studi di proteomica, offre un notevole aiuto alla chimica farmaceutica per la sintesi di nuovi farmaci in tempi relativamente più brevi e con costi meno elevati.
Il procedimento di sintesi di un farmaco consta di numerose fasi, le quali mirano a garantire l’immissione sul mercato di un prodotto farmaceutico sicuro e con eventuali pochi effetti collaterali. Pertanto, il tempo che intercorre tra le fasi iniziali di produzione di un farmaco e la vendita di quest’ultimo non è inferiore ai quindici anni.
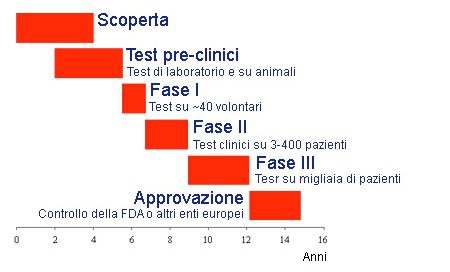
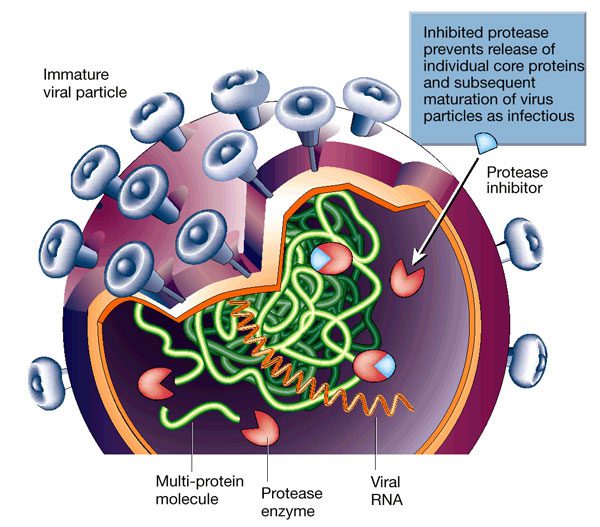
In primo luogo, è, ovviamente, essenziale l’identificazione della molecola bersaglio, ovverosia della molecola biologica necessaria per la sopravvivenza e per la proliferazione dell’agente eziologico della malattia oggetto di studio. In seguito all’identificazione della molecola bersaglio del futuro farmaco, è altrettanto fondamentale individuare uno o più composti guida, cioè uno o più composti che legano il sito attivo della molecola bersaglio e ne inibiscano l’attività. Un composto guida, dunque, poiché inibisce l’attività della molecola bersaglio, la quale è essenziale per la sopravvivenza del patogeno, può bloccare la proliferazione di quest’ultimo e distruggerlo. Per chiarire meglio le definizioni di molecola bersaglio e composto guida sopra descritte, si consideri, come esempio, l’enzima HIV proteasi, il quale è una proteina prodotta dal virus umano dell’immunodeficienza (HIV, Human Immunodeficiency Virus), l’agente responsabile della sindrome da immunodeficienza acquisita (AIDS, Acquired Immune Deficiency Syndrome). L’enzima proteasi dell’HIV può interagire con le catene laterali di taluni amminoacidi contenuti in substrati polipeptidici che fungono da siti di clivaggio per la proteina virale e, tramite tale interazione, può processare il precursore poliproteico espresso dai geni gag e gag – pol e produrre proteine mature e funzionali – si ricordi che il gene gag (group – specific antigen) codifica per le proteine strutturali del core del virus e che il geni gag – pol sono coespressi in una poliproteina la quale viene processata a sintetizzare una proteina di 55 KDa e tre enzimi essenziali per l’attività virale: proteasi, transcrittasi inversa e integrasi.
In tempi recenti, sono stati prodotti numerosi inibitori della proteasi dell’HIV. Alcuni inibitori detti competitivi possono mimare la presenza del substrato polipeptidico nel sito attivo della proteasi impedendo l’effettiva attività dell’enzima, altri, invece, possono modificare covalentemente lo stesso sito attivo della proteasi impedendone il legame con i propri substrati.
Poiché è l’interazione tra il composto guida e la molecola bersaglio l’evento principale per la determinazione di un nuovo farmaco, numerose tecniche computazionali di docking (dall’inglese, interazione, legame) molecolare mirano allo studio della modalità di interazione tra due molecole con struttura nota. I principi fondamentali che un bioinformatico deve considerare nella determinazione della struttura del complesso che si forma tra due molecole sono i seguenti: come nell’uso di programmi bioinformatici per lo studio del folding (dall’inglese, ripiegamento) proteico, il complesso molecola bersaglio – composto guida deve dare origine ad una conformazione stabile, a cui corrisponde, cioè, il più basso valore di energia. L’altra considerazione da non trascurare e, perciò, non meno importante della funzione energia, riguarda la gestione della flessibilità sia del composto guida sia della molecola bersaglio: il bioinformatico può scegliere tra un approccio tipo chiave – serratura, mediante il quale la molecola bersaglio può assumere una struttura proteica rigida a cui si aggancia il composto guida flessibile ed un approccio di docking con adattamento indotto, il quale consente l’assunzione di più gradi di flessibilità sia da parte della molecola bersaglio sia da parte del composto guida.
Nel corso degli anni, sono stati ideati ed utilizzati diversi programmi informatici di docking: FTDock abbinato a MultiDock, gli algoritmi HEX, DOCK, Hammerhead, GOLD, FlexX. In tempi recenti, per ridurre il numero di complessi da considerare e diminuire i tempi di ricerca per la scoperta di un composto guida – e, quindi, di un farmaco – che leghi ed inibisca una molecola bersaglio, è stato utilizzato l’algoritmo SLIDE, il quale, mediante tecniche di indicizzazione dei database, scarta tutti i composti guida la cui probabilità di legare il sito attivo della molecola bersaglio è notevolmente bassa.
Maria Chiara Langella
Fonti: Fondamenti di bioinformatica. Dan E. Krane, Michael L. Raymer. PEARSON Benjamin Cummings. Edizione italiana a cura di Eugenia Polverini. 2007. Pagine 193 – 197.
http://www.scuolafn.unicz.it/docenti/alcaro/f1/aa_05_06/tesine/rogato.pdf
http://www.sunhope.it/I%20RETROVIRUS%20UMANI.pdf
Immagine in evidenza da https://www.disva.univpm.it/content/laboratorio-di-modeling-molecolare-e-bionanotecnologie
Immagine 1 da https://www.bioblog.it/2007/11/26/farmaci-fasi-ricerca-clinica-test-clinici/20071811
Immagine 2 da https://www.liquidarea.com/2009/04/meccanismo-di-azione-degli-inibitori-della-proteasi/