Introduzione
La tassonomia è un principio organizzativo della biologia che si basa sulle relazioni evolutive tra gli organismi. In particolare, lo sviluppo di una tassonomia batterica è spesso ostacolato dall’incapacità di poter ottenere colture pure di alcune tipologie batteriche e, in misura minore, dall’uso dei fenotipi per guidare la classificazione. Con l’avvento di nuove tecniche di sequenziamento e la nascita di banche dati in globale condivisione, la filogenesi fenotipica è stata sostituita dalla cosiddetta filogenesi molecolare. L’analisi comparativa di sequenze relative agli RNA ribosomiali (Fig. 1) o dei geni corrispondenti può essere considerato come l’approccio più attuale sia per la ricostruzione della filogenesi microbica sia per l’identificazione batterica.
Diverse indagini hanno evidenziato che gli alberi basati su alcune sequenze di rRNA possono riflettere in maniera globalmente corretta la storia dei corrispondenti organismi. Tuttavia, considerati tre/quattro miliardi di anni di evoluzione, è anche necessario tener conto del fatto che il contenuto informativo di tali sequenze non è sempre ben conservato e utile a ricostruire la storia filogenetica in maniera precisa e dettagliata. Pertanto, solo un’attenta manipolazione dei dati permette un utilizzo ragionevole delle informazioni filogenetiche limitate. L’attenta analisi consiste in un corretto allineamento, un’attenta analisi della variabilità posizionale, del tipo e del tasso di cambiamento testando varie selezioni di dati. Risulta fondamentale, inoltre, applicare metodi alternativi di alberatura accompagnati all’esecuzione di test di confidenza. La metodologia attuale più affascinante è senza dubbio l’ibridazione cellulare in situ, attraverso la quale singole cellule batteriche possono essere identificate all’interno di campioni complessi.
RNA e la scelta delle sequenze utili all’analisi
Nell’ambito della filogenesi microbica, le molecole di rRNA più grandi (16S e 23S rRNA) sono utili come misura delle relazioni filogenetiche poiché possiedono tutti i prerequisiti associati alle molecole marcatrici filogenetiche. Un primo aspetto fondamentale che spesso non viene considerato nelle indagini relative al 16S rRNA è l’utilizzo di sequenze del gene più lunghe di 1.300 nucleotidi che permettono indagini ad alta produttività ed affidabilità. Un secondo aspetto importante per le analisi di tipo filogenetico, indipendentemente dall’ambito in cui essa viene applicata, riguarda la scelta di un marcatore filogenetico utile e valido.
Tra le caratteristiche di queste molecole (o geni) si ritrova la conservazione funzionale, la distribuzione ubiquitaria, le dimensioni sufficienti e la presenza simultanea di elementi di struttura che sono evolutivamente conservati. Talvolta, possono essere considerate anche sequenze variabili in modo da osservare le più recenti variazioni/distanze filogenetiche tra i microrganismi in esame. Un esempio dei profili di conservazione (Fig. 2) è quello relativo a quattro diversi livelli filogenetici, cioè il dominio Bacteria, i batteri Gram-positivi con un basso contenuto di DNA G + C, il gruppo filogenetico che combina i bacilli, lattobacilli, lattococchi, streptococchi, listeriae e i membri del genere Staphylococcus.
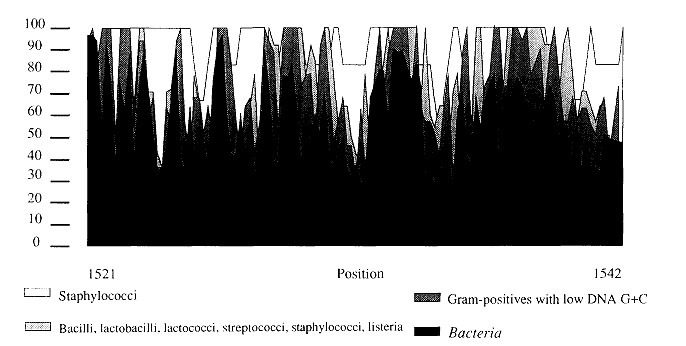
L’immagine mostra i gradi di conservazione all’interno di una precisa parte della sequenza. I profili sono stati calcolati definendo la base più frequente in ogni singola posizione all’ interno di ogni singolo gruppo.
Allineamento delle sequenze
La regola fondamentale riguarda la collocazione delle posizioni omologhe infatti, queste devono essere disposte in colonne comuni ed in un corretto allineamento. Con i marker filogenetici, che contengono essenzialmente sequenze conservate, l’allineamento risulta una procedura semplice che può essere eseguita in modo affidabile utilizzando un software di allineamento multiplo come MEGA (Molecular Evolutionary Genetics Analysis). Nel caso delle sequenze di amminoacidi, le ambiguità di allineamento possono spesso essere risolte organizzando gli aminoacidi sulla base delle loro caratteristiche biochimiche. Per la selezione delle sequenze da utilizzare, esistono banche dati completamente accessibili al pubblico. Tra quelle di maggior rilievo vi sono NCBI e RDB, quindi il “Ribosomal Database Project” nato con l’intento di rendere fruibili al pubblico vari tipi di allineamenti che vengono costantemente aggiornati da specialisti.
Costruzione degli alberi
L’elemento chiave per la visualizzazione dei dati filogenetici si trova nella realizzazione di alberi additivi nei quali la ramificazione indica il percorso dell’evoluzione e la lunghezza (elemento additivo) dei rami indica la distanza filogenetica. Al termine, gli alberi sono mostrati in vari formati quindi radiale (Fig. 3) o sotto forma di dendrogramma (Fig. 4).
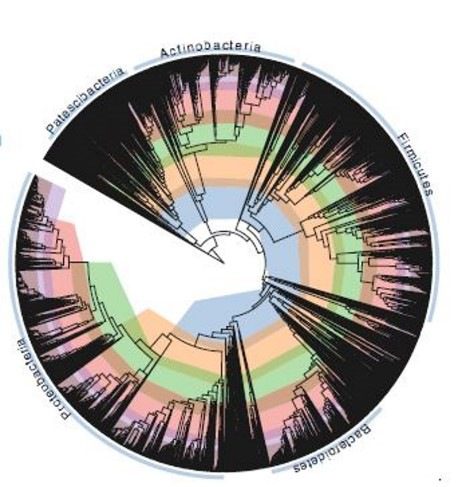
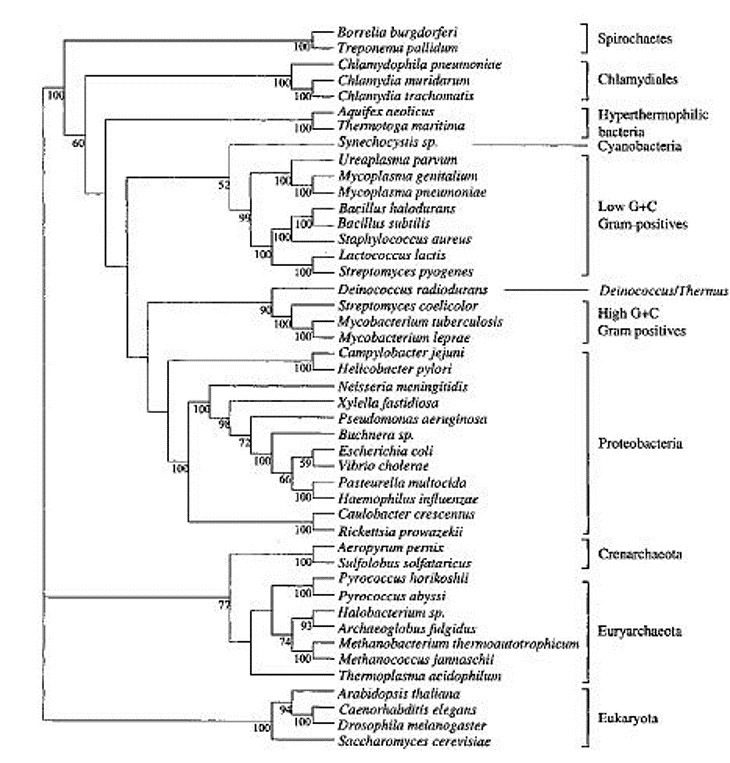
Comunemente, si riconoscono tre approcci principali per la ricostruzione di un albero: matrice di distanza, massima parsimonia e metodi di massima verosimiglianza. Date le possibili incongruenze dei dati di sequenza ed il fatto che i diversi metodi di alberatura operano secondo regole o presupposti diversi, è altamente raccomandato di non fidarsi dei risultati ma di eseguire test di fiducia sugli alberi ottenuti. In pratica, si considera come buona prassi quella di mettere un 1-2% si sfocatura intorno ai singoli rami per arrivare ad una prima stima del possibile errore.
Una valida alternativa: la filogenesi proteica
Una particolare metodica usata per la classificazione dei microrganismi è la filogenesi proteica concatenata. Tale approccio rimuove conservativamente i gruppi polifiletici e normalizza i gradi tassonomici sulla base della relativa divergenza evolutiva. Questo diverso approccio, che viene considerato come “tassonomia GTDB“, è pubblicamente disponibile sul sito web del Genome Taxonomy Database. Il confronto tra questo nuovo approccio basato sull’analisi proteica con i dati tassonomici presenti nella piattaforma pubblica NCBI (Fig. 5), ha evidenziato notevoli differenze relative alla tassonomia e sottolineato il fatto che il tipo di approccio può modificare notevolmente i risultati tassonomici e la classificazione dei microrganismi considerati.

Con l’utilizzo della classificazione GTDB è stata proposta una tassonomia batterica che copre 94.759 genomi batterici, compresi 13.636 (14,4%) provenienti da organismi non coltivati (ottenuti grazie a metagenoma assemblato o genomi di una singola cellula). Tale tipo di tassonomia si presenta come ampiamente congruente con i tassi di sostituzione delle filogenesi dedotte utilizzando vari marcatori e metodi di inferenza a massima verosimiglianza.
Gli ostacoli della filogenesi procariotica
Si possono distinguere due grandi ostacoli: primo, la frequenza con cui si verificano i trasferimenti laterali e la paralogia nascosta; in secondo luogo, la perdita di segnale filogenetico per i rami profondi. Per aggirare questi ostacoli, sono state utilizzate diverse strategie nello studio della filogenesi procariotica, tra queste alcune evitano di considerare le informazioni contenute nelle sequenze perché possono essere fuorvianti quindi considerano un gene come un carattere a sé stante. Questi metodi sembrano essere molto sensibili ai trasferimenti laterali e alla perdita di geni, risolvendo errori legati a questi fenomeni. Altri metodi, invece, cercano di aumentare il segnale filogenetico concatenando i geni tra loro.
Conclusioni
Considerate le diverse varianti con cui può essere applicata la classificazione filogenetica e il rapido sviluppo di nuovi metodi di identificazione, risulta fondamentale un processo di coordinamento mondiale relativo all’organizzazione dei dati con l’obiettivo di definire degli standard minimi per l’elaborazione e l’interpretazione ottimale. Attualmente, sono già note banche dati contenenti sequenze di rRNA allineate da specialisti, ma la frequenza e la velocità di aggiornamento sono molto inferiori rispetto al numero di sequenze in rapida crescita. L’approccio associato alle sequenze di rRNA rende plausibile ipotizzare che in futuro tutte le specie batteriche saranno analizzate filogeneticamente utilizzando questo metodo fino ad arrivare, probabilmente, ad una classificazione genealogica corretta, inequivocabile ed universalmente valida.
Fonti
- https://www.megasoftware.net/
- https://www.ncbi.nlm.nih.gov/
- http://gtdb.ecogenomic.org/
- https://rdp.cme.msu.edu/
- David S. Goodsell, RCSB Protein Data Bank – Molecule of the Month at the RCSB Protein Data Bank
- Daubin V, Gouy M, Perrière G. A phylogenomic approach to bacterial phylogeny: evidence of a core of genes sharing a common history. Genome Res. 2002 Jul;12(7):1080-90. doi: 10.1101/gr.187002. PMID: 12097345; PMCID: PMC186629.
- Gupta RS, Griffiths E. Critical issues in bacterial phylogeny. Theor Popul Biol. 2002 Jun;61(4):423-34. doi: 10.1006/tpbi.2002.1589. PMID: 12167362.
- Ludwig W, Schleifer KH. Bacterial phylogeny based on 16S and 23S rRNA sequence analysis. FEMS Microbiol Rev. 1994 Oct;15(2-3):155-73. doi: 10.1111/j.1574-6976.1994.tb00132.x. PMID: 7524576.
- Ludwig W, Strunk O, Klugbauer S, et al. Bacterial phylogeny based on comparative sequence analysis. Electrophoresis. 1998 Apr;19(4):554-568. DOI: 10.1002/elps.1150190416.
- Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil PA, Hugenholtz P. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. 2018 Nov;36(10):996-1004. doi: 10.1038/nbt.4229. Epub 2018 Aug 27. PMID: 30148503.
- Yarza, P., Yilmaz, P., Pruesse, E. et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12, 635–645 (2014). https://doi.org/10.1038/nrmicro3330