L’acrocefalopolidattilia, conosciuta anche come sindrome di Apert, è una rara e complessa malattia genetica che colpisce lo sviluppo craniofacciale e scheletrico. Questa patologia è caratterizzata da una combinazione di anomalie craniche, facciali e delle estremità, creando sfide sia per la salute che per la funzionalità dei pazienti. In questo articolo, esploreremo in dettaglio le cause, i sintomi, la diagnosi, la prevenzione e le opzioni di trattamento per questa rara condizione.
Cause dell’Acrocefalopolidattilia
L’acrocefalopolidattilia è causata da mutazioni genetiche nel gene FGFR2, che è responsabile della regolazione della crescita e dello sviluppo delle ossa e dei tessuti connettivi nel corpo umano. Queste mutazioni influenzano negativamente lo sviluppo craniofacciale e delle estremità durante la gestazione, portando alla formazione anomala del cranio e delle dita delle mani e dei piedi.
Secondo il Centro per il Controllo e la Prevenzione delle Malattie, l’acrocefalopolidattilia colpisce circa 1 su 65.000-88.000 nati vivi. La malattia non mostra preferenze di genere o gruppo etnico e può verificarsi in famiglie senza storia genetica della condizione.
Sintomi Caratteristici dell’Acrocefalopolidattilia
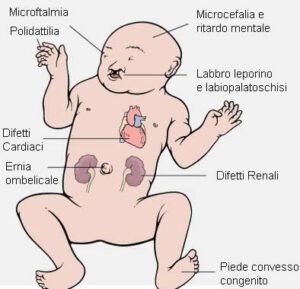
I sintomi dell’acrocefalopolidattilia sono ampiamente riconoscibili e includono:
- Cranio e Faccia Anomali: I pazienti spesso presentano un cranio deformato, chiamato craniosinostosi, che porta a una forma irregolare del cranio e degli occhi sporgenti. Il volto può apparire piatto e allungato, con un naso piatto e largo.
- Dita Fuse: Le estremità sono colpite con sindattilia, dove le dita delle mani e dei piedi possono essere fuse insieme. Questo può compromettere la normale funzionalità delle mani e dei piedi.
- Problemi Dentari: Problemi dentari come il morso incrociato sono comuni, a causa della conformazione irregolare della mascella.
- Problemi Respiratori e dell’Udito: Le anomalie craniche possono causare difficoltà respiratorie e problemi all’udito nei pazienti affetti.
Diagnosi e Approccio Clinico
La diagnosi dell’acrocefalopolidattilia si basa su esami clinici approfonditi e imaging radiologici come la radiografia del cranio e delle mani. La valutazione genetica è essenziale per confermare la presenza di mutazioni nel gene FGFR2.
Il trattamento richiede un approccio multidisciplinare, coinvolgendo chirurghi cranio-maxillo-facciali, ortopedici, odontoiatri e terapisti occupazionali. L’intervento chirurgico è spesso necessario per correggere le anomalie craniche e delle estremità, migliorando la funzionalità e l’aspetto estetico.
Prevenzione e Trattamento
Poiché l’acrocefalopolidattilia è una malattia genetica, la prevenzione primaria si concentra sulla consulenza genetica per le coppie con una storia familiare della sindrome. La consulenza genetica aiuta a comprendere i rischi di trasmissione ai figli e a esplorare le opzioni disponibili.
Il trattamento mira a migliorare la qualità della vita del paziente. Le opzioni terapeutiche includono:
- Chirurgia Correttiva: Gli interventi chirurgici correttivi sono fondamentali per migliorare la forma del cranio, il posizionamento degli occhi e delle orecchie, nonché per separare le dita fuse.
- Terapia Occupazionale e Fisica: I terapisti aiutano i pazienti a sviluppare abilità motorie e funzionalità delle mani e dei piedi attraverso esercizi mirati.
- Cura Dentale: Gli specialisti dentali gestiscono problemi come il morso incrociato e altre anomalie dentali.
Conclusioni
L’acrocefalopolidattilia è una complessa malattia genetica che richiede un approccio clinico integrato per il trattamento e la gestione. La diagnosi precoce, l’intervento chirurgico tempestivo e il supporto terapeutico possono migliorare notevolmente la qualità della vita dei pazienti affetti da questa sindrome. La consulenza genetica svolge un ruolo cruciale nella prevenzione, aiutando le famiglie a prendere decisioni informate per il futuro.