Caratteristiche dell’atrofia muscolare spinale
L’atrofia muscolare spinale (SMA) è una malattia neurodegenerativa rara, caratterizzata da una progressiva degenerazione dei motoneuroni spinali e del tronco encefalico. Si manifesta con atrofia e conseguente indebolimento dei muscoli scheletrici, e difficoltà motorie.
Esistono cinque differenti forme di atrofia muscolare spinale: il tipo 0, il tipo 1, il tipo 2, il tipo 3 e il tipo 4.
L’atrofia muscolare spinale può causare la morte del paziente di età giovane. Le forme più gravi della malattia, infatti, compromettono l’efficienza dei muscoli respiratori, responsabili di episodi di insufficienza respiratoria o polmonite dall’esito mortale.
Eziologia e patogenesi dell’atrofia muscolare spinale
Il primo caso di atrofia muscolare spinale (SMA) fu accertato in un adulto nel 1850, da due medici francesi François-Amilcar Aran e Guillaume Duchenne. Successivamente fu individuata una variante più grave in un bambino dai tedeschi Guido Werdnig e Johann Hoffmann.
L’atrofia muscolare spinale è dovuta a mutazioni a carico del gene coinvolto nello sviluppo del sistema nervoso, chiamato gene del motoneurone 1 di sopravvivenza (SMN1), il quale codifica per una proteina detta proteina del motoneurone di sopravvivenza (SMN), che ha un ruolo fondamentale per il mantenimento e la funzionalità di tutti i motoneuroni. La proteina SMN è localizzata sul cromosoma 5(5q12-13).
Con un livello non adeguato della proteina SMN, i motoneuroni del midollo spinale degenerano, impedendo ai muscoli di ricevere i segnali corretti dal cervello.

I pazienti affetti da atrofia muscolare spinale, presentano un secondo gene, chiamato SMN2 (gene del motoneurone 2 di sopravvivenza), che codifica per una forma accorciata della proteina SMN. Il gene SMN2 ha una struttura simile al gene SMN1, ma solo una piccola quantità della proteina SMN che produce è completamente funzionale. Il numero di copie del gene SMN2 nei pazienti affetti da SMA può variare e può portare a forme più o meno gravi.
Classificazione
L’atrofia muscolare spinale è classificata in cinque tipi:
| Tipo | Età esordio | Caratteristiche |
| Tipo 0 | Prenatale | Forma più grave in assoluto. Si manifesta prima ancora della nascita con ridotta mobilità del feto. Dopo la nascita, i piccoli malati sopravvivono poche settimane, anche con il supporto respiratorio. |
| Tipo 1 | 0-6 mesi | Chiamata anche malattia di Werdnig-Hoffmann. E’ la forma più severa e comune; compare entro il sesto mese di vita. Causa la morte già nei primi anni di vita, raramente durante l’adolescenza. Il decesso avviene per un’insufficienza respiratoria o per infezione polmonare. |
| Tipo 2 | 7-18 mesi | Chiamata anche malattia di Dubowitz, esordisce tra i 7 e i 18 mesi di vita. L’aspettativa di vita per chi ne affetto è maggiore rispetto al tipo 1: infatti i pazienti, raggiungono l’età adulta. |
| Tipo 3 | >12 mesi | Detta anche malattia di Kugelberg-Welander, si manifesta tipicamente dopo i 18 mesi di vita. Comporta disabilità importanti, ma non pregiudica l’aspettativa di vita. |
| Tipo 4 | Età adulta | Esordisce in età adulta ed è la forma meno grave; ha un decorso molto lento. In genere, non è responsabile di problemi respiratori ed è associata a un’aspettativa di vita normale. |
Segni e sintomi dell’atrofia muscolare spinale
L’atrofia muscolare spinale si caratterizza da diversi sintomi che possono includere:
- Ipotonia;
- Atrofia muscolare;
- Debolezza muscolare;
- Scoliosi;
- Tremori alle dita delle mani;
- Paralisi;
- Disfunzioni cardiache;
- Fascicolazioni (ovvero contrazioni) della lingua;
- Difficoltà a raggiungere le tappe dello sviluppo;
- Difficoltà a stare seduti, in piedi e a camminare;
- Perdita di forza dei muscoli respiratori con conseguente tosse debole, pianto debole (nei neonati), accumulo di secrezioni nei polmoni o nella gola.
In base al tipo di atrofia muscolare spinale alcuni sintomi possono essere più o meno debilitanti:
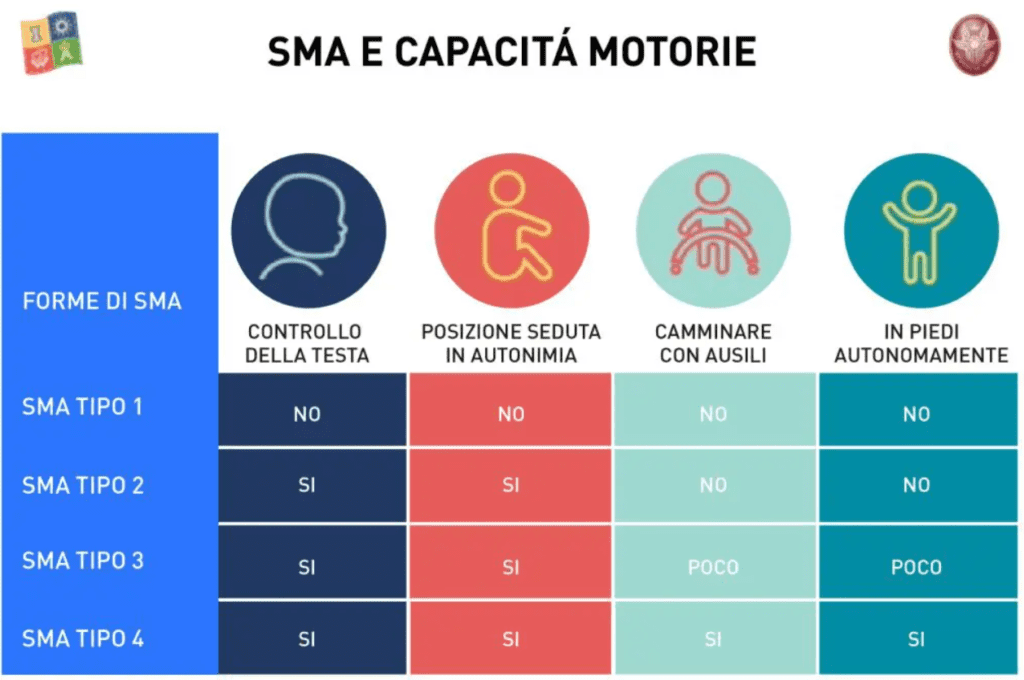
Epidemiologia
E’ una malattia a trasmissione ereditaria autosomica recessiva, ciò significa che un neonato, per essere a rischio, deve ereditare 1 gene SMN1 mutato da ciascun genitore. Se un neonato eredita 1 solo gene SMN1 mutato, viene considerato un “portatore”, ma non manifesta i sintomi dell’atrofia muscolare spinale.

L’atrofia muscolare spinale presenta un’incidenza annuale di 1 caso ogni 10.000 nati.
Diagnosi
L’atrofia muscolare spinale viene diagnosticata mediante test genetici effettuati su campioni di sangue, attraverso la tecnica MLPA, che rilevano mutazioni a carico di SMN1/SMN2.
Prima di effettuare il test genetico per l’identificazione della SMA, viene eseguito un esame obiettivo e un’anamnesi sospetti, che vanno a rilevare sintomi tipici (indebolimento e mollezza dei muscoli; contrazioni muscolari improvvise; riflessi tendinei ridotti o assenti) e una storia familiare riconducibili all’atrofia muscolare spinale.
E’ inoltre possibile diagnosticare l’atrofia muscolare spinale nel periodo prenatale tramite test genetico su un campione di cellule fetali, ottenute tramite amniocentosi o villocentosi.
Infine durante la diagnosi per l’atrofia muscolare spinale, potrebbero trovare impiego esami come l’elettromiografia o la biopsia muscolare.
Terapia
L’approccio terapeutico più diffuso tra i pazienti con atrofia muscolare spinale è il trattamento sintomatico, ovvero basato su trattamenti che alleviano la sintomatologia e le complicanze, e che mirano a migliorare il tenore di vita dei malati. I trattamenti si classificano in:
- Supporto respiratorio: utilizzo di maschere per la ventilazione, intubazione orotracheale e tracheostomia utili per la respirazione e per prevenire le infezioni polmonari.
- Supporto alla nutrizione: utilizzo di sondino naso gastrico, intervento di gastrostomia e seguire una dieta equilibrata alle esigenze del paziente per agevolare la deglutizione e masticazione del cibo.
- Supporto fisioterapico: per migliorare, la flessibilità dei muscoli e rendere meno rigide le articolazioni.
- Supporto ortopedico: per migliorare la scoliosi si utilizza un busto ortopedico, quando la deformazione è lieve, oppure si può optare per un intervento chirurgico di fusione spinale, quando la malformazione è grave.
Esistono anche farmaci specifici contro la SMA. Quelli attualmente utilizzati sono Nusinersen e Risdiplam: il primo agisce correggendo in corso la produzione aberrante della proteina SMN; il secondo incrementa i livelli di produzione di SMN, tentando anche di mantenerli a una quota adeguata alle esigenze dell’organismo umano.
Nel 2019 l’FDA ha approvato una tecnica, basata sulla strategia della terapia genica, che permette di correggere la mutazione responsabile della SMA e proteggere i motoneuroni dalla progressiva degenerazione. La tecnica è denominata Zolgensma, basata sull’impiego di un virus-vettore capace di inserire nel DNA all’interno dei motoneuroni di un malato una copia normale del gene SMN1/SMN2.
Fonti
- https://it.wikipedia.org/wiki/
- https://www.my-personaltrainer.it/salute-benessere/.html
- https://en.wikipedia.org/wiki/
- https://www.osservatoriomalattierare.it/malattie-rare/sma
- https://www.microbiologiaitalia.it/virologia/sma-pediatricafda-approva-zolgensmaterapia-genica-a-base-di-virus-adeno-associati/
Fonti immagini
- Immagine in evidenza: https://www.everydayhealth.com/genetic-diseases/doctor-questions-spinal-muscular-atrophy/
- Figura 1:https://care.togetherinsma.it/it_IT/home/introduction-to-sma/smn1-gene.html
- Figura 2: https://biomedicalcue.it/meyer-firenze-terapia-genica-neonato-sma1/29770/
- Figura 3: https://care.togetherinsma.it/it_IT/home/introduction-to-sma/smn1-gene.html