La Beta Talassemia, conosciuta anche come Talassemia β, è una patologia ematologica ereditaria caratterizzata da un’anomala produzione dell’emoglobina β, una delle due catene proteiche che costituiscono l’emoglobina, responsabile del trasporto dell’ossigeno nei globuli rossi. Questa condizione genetica può causare gravi disturbi a livello ematico e comportare una vasta gamma di sintomi e complicanze. In questo articolo, esploreremo le cause, i sintomi, le diagnosi, le opzioni terapeutiche e gli aspetti prognostici della Beta Talassemia.
Cause e Tipi di Beta Talassemia
La Beta Talassemia è una malattia ereditaria causata da mutazioni genetiche nel gene HBB, responsabile della sintesi dell’emoglobina β. Queste mutazioni possono portare a un’anomala produzione o una quantità insufficiente di emoglobina beta, influenzando la capacità dei globuli rossi di trasportare ossigeno. A seconda del tipo e del numero di mutazioni presenti, la Beta Talassemia può essere classificata in diverse forme:
- β Talassemia Maggiore (Talassemia Major): Questa forma si manifesta quando entrambi i genitori trasmettono una copia del gene mutato al loro figlio. È la forma più grave di Beta Talassemia, con sintomi significativi e necessità di trattamento medico regolare per tutta la vita.
- β Talassemia Intermedia (Talassemia Intermedia): In questa variante, il paziente eredita una copia del gene mutato da uno dei genitori. I sintomi possono variare ampiamente da lievi a moderati, e talvolta è possibile gestire la condizione senza trasfusioni regolari di sangue.
- β Talassemia Minor (Talassemia Minor): Questo tipo si verifica quando il paziente eredita una sola copia del gene mutato da uno dei genitori. Le persone con Beta Talassemia minore possono essere portatori sani e spesso non manifestano sintomi significativi.
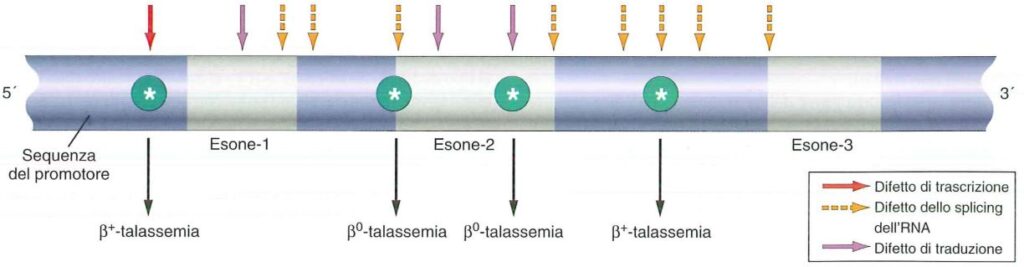
Sintomi e Complicanze della Beta Talassemia
I sintomi della β Talassemia possono variare notevolmente in base alla gravità della malattia e al tipo di mutazioni presenti. Tuttavia, alcuni sintomi comuni includono:
- Anemia: I pazienti con Beta Talassemia spesso presentano anemia cronica, che si manifesta con stanchezza, pallore della pelle e debolezza.
- Problemi ossei: La Beta Talassemia può causare deformità ossee, inclusa l’ingrandimento della milza e dell’epatosplenomegalia.
- Crescita ritardata: Nei bambini affetti da Beta Talassemia, la malattia può influenzare negativamente la crescita e lo sviluppo.
- Ittero: L’accumulo di bilirubina a causa della rottura dei globuli rossi può causare ittero (ingiallimento della pelle e degli occhi).
- Problemi cardiaci: In alcuni casi, la Beta Talassemia può provocare insufficienza cardiaca, a causa dell’eccesso di ferro accumulato a seguito delle trasfusioni di sangue.
- Formazione di calcoli: Il sovraccarico di ferro nel sangue può portare alla formazione di calcoli biliari.
Diagnosi e Screening
La diagnosi precoce della β Talassemia è fondamentale per garantire una gestione efficace e una migliore qualità della vita per i pazienti. Gli screening prenatale e neonatale sono essenziali per individuare la presenza di questa patologia e consentire un intervento tempestivo.
I test diagnostici per la Beta Talassemia includono:
- Emocromocitometria (CBC): Questo esame misura i livelli di emoglobina e il numero di globuli rossi nel sangue per rilevare l’anemia.
- Elettroforesi dell’emoglobina: Questo test separa e identifica le diverse varianti di emoglobina presenti nel sangue, inclusa l’emoglobina anomala tipica della Beta Talassemia.
- Test genetici (DNA): I test genetici possono confermare la presenza di mutazioni specifiche associate alla Beta Talassemia.
Trattamento e Gestione
La gestione della Beta Talassemia dipende dalla gravità della condizione. Alcune opzioni di trattamento comuni includono:
- Transfusioni di sangue: I pazienti con Beta Talassemia maggiore possono necessitare di trasfusioni regolari per mantenere livelli adeguati di emoglobina e prevenire complicazioni legate all’anemia.
- Chelazione del ferro: A causa delle trasfusioni di sangue frequenti, può accumularsi eccesso di ferro nell’organismo, che viene rimosso mediante trattamenti di chelazione.
- Trapianto di cellule staminali: Nei casi più gravi, un trapianto di cellule staminali da un donatore compatibile può essere considerato come terapia curativa.
Conclusioni
La Beta Talassemia è una patologia ematologica ereditaria che può causare una serie di sintomi e complicanze. La diagnosi precoce e il trattamento adeguato sono cruciali per migliorare la qualità della vita dei pazienti affetti da questa malattia. Gli screening genetici e i test diagnostici sono strumenti importanti per individuare tempestivamente la presenza della Beta Talassemia e consentire un intervento mirato.
L’avanzamento della ricerca scientifica e la consapevolezza pubblica possono contribuire a migliorare le opzioni terapeutiche e la prognosi dei pazienti affetti da questa patologia. Sostenere la ricerca medica e promuovere la prevenzione sono passi fondamentali verso una migliore gestione della Beta Talassemia e altre malattie ematologiche ereditarie.
Oltre ogni ostacolo. La Talassemia: una storia personale e collettiva a Ferrara
Fonti
- Cappellini MD, Cohen A, Piga A, et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with β-thalassemia. Blood. 2006;107(9):3455-3462.
- Taher AT, Musallam KM, Cappellini MD. β-Thalassemias. N Engl J Med. 2021;384(8):727-743.
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5:11.
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61-76.
- Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331-4336.
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480-487.
- CRISPR-Cas9 per la talassemia: primo caso in Italia