Deficit di Adenosina Deaminasi
Il deficit di Adenosina Deaminasi (ADA-SCID) è una patologia rara, di origine genetica, che appartiene al gruppo delle immunodeficienze severe combinate (SCID), malattie che causano una grave compromissione del sistema immunitario, esponendo il soggetto a infezioni ricorrenti, con esito spesso fatale.
Eziologia e trasmissione del deficit di Adenosina Deaminasi (ADA-SCID)
La patologia è causata da una mutazione con perdita di funzione del gene ADA, localizzato sul cromosoma 20, che codifica per l’enzima Adenosina Deaminasi (ADA), che svolge un ruolo cruciale per il funzionamento del sistema immunitario e la maturazione dei linfociti T. Più nello specifico, l’enzima interviene nel catabolismo delle purine, catalizzando la conversione dell’adenosina e della 2’-deossiadenosina, rispettivamente in inosina e 2’-deossinosina.
![gura 1 - Ruolo dell'enzima nel pathway catabolico delle purine. [fonte: frontiersin.org]](https://www.microbiologiaitalia.it/wp-content/uploads/2022/04/fimmu-07-00314-g001.jpg)
La 2’-deossiadenosina agisce come inibitore dell’enzima S-adenosilomocisteina idrolasi (SAH), e in presenza del gene ADA mutato, si verifica un suo notevole incremento intracellulare, portando di conseguenza all’accumulo di SAH, che inibisce una serie di meccanismi cellulari essenziali per il differenziamento e le funzioni effettrici dei linfociti T. Si assiste inoltre ad un aumento dei livelli citoplasmatici di ATP, che regola negativamente l’enzima ribonucleotide reduttasi (RR), causando difetti nei sistemi di replicazione e riparazione del DNA.
D’altra parte, l’aumento di livelli di adenosina, conduce all’accumulo di AMP ciclico (cAMP), una molecola segnale che inibisce l’attività dei linfociti e di conseguenza la risposta immunitaria del soggetto.
L’ADA-SCID si trasmette con ereditarietà autosomica recessiva, questo vuole dire che il soggetto, per essere affetto, deve possedere entrambe le copie mutate del gene ADA. In presenza di genitori entrambi portatori, il figlio ha il 25% di manifestare la patologia, ovvero di ereditare entrambi gli alleli mutati.
![Figura 2 - Meccanismo di ereditarietà dell'ADA-SCID. [fonte: osmosis.org]](https://www.microbiologiaitalia.it/wp-content/uploads/2022/04/image-4.jpg)
Aspetti clinici dell’ADA-SCID
Nella maggior parte dei casi, la patologia si presenta con infezioni ricorrenti, spesso causate da microrganismi che normalmente sono innocui per un l’uomo. Nei soggetti affetti invece, si possono verificare infezioni che, in presenza di un sistema immunitario notevolmente indebolito, hanno un decorso molto aggressivo, portando a morte precoce. Nel 10-15% dei casi la patologia si manifesta più tardivamente, a 6-24 mesi e una piccola percentuale di casi invece presenta una forma ad esordio tra i 4 anni e l’età adulta.
Anche patogeni come Virus-Varicella Zoster, o persino Rhinovirus, agente del comune raffreddore, possono avere una prognosi infausta, tanto che questi soggetti sono costretti a vivere in ambienti totalmente asettici e isolati per evitare il rischio di infettarsi (i cosiddetti “bambini bolla”, definizione oggi non più in uso). La maggior parte delle infezioni sono di microorganismi opportunisti (batteri e funghi) e interessano le alte vie aeree, provocando sinusiti, otiti e faringiti ricorrenti. Si possono inoltre verificare anche manifestazione extra-respiratorie, che interessano il sistema digerente e la pelle, causando rispettivamente, diarrea persistente e dermatite severa.
Il deficit di adenosina deaminasi, contrariamente a quanto si possa pensare, non è una patologia che riguarda solo il sistema immunitario, ma interessa anche il sistema nervoso (deficit dello sviluppo neurale, disturbi del comportamento e sordità neurosensoriale), il sistema scheletrico (rallentamento della crescita ossea) e altre manifestazioni epatiche ed ematologiche.
Diagnosi e terapia
La diagnosi di ADA-SCID viene condotta a partire dall’osservazione clinica del soggetto e in seconda istanza si eseguono esami di laboratorio per conferma. I parametri laboratoristici che vengono presi in considerazione alla nascita sono:
- Linfopenia: in presenza di livello di linfociti inferiore ai 500 /µl (nei neonati i valori di riferimento sono compresi tra i 2000 e i 5000/µl
- Bassi livelli di immunoglobuline sieriche (Ig)
- Elevati livelli plasmatici di deossiadenosina (dAdo) e adenosina
- Ridotta attività dell’enzima S-Adenosilomocisteina idrolasi negli eritrociti (<5%).
Successivamente si esegue l’analisi genetica per accertare la presenza di mutazione in omozigosi del gene ADA, che può essere eseguita in due modi:
- Indagine del singolo gene: si esegue l’analisi del gene ADA in cui si studiano duplicazioni/delezioni.
- Utilizzo di un pannello multigenico: Si esegue l’analisi del gene ADA e di altri geni di interesse che possono mettere l’ADA-SCID in diagnosi differenziale con altre patologie che presentano fenotipi clinici simili.
Le opzioni terapeutiche attualmente in uso hanno avuto un’evoluzione incredibile nel corso dell’ultimo ventennio. In passato l’unico trattamento terapeutico disponibile era il trapianto di midollo osseo, effettuabile solo in presenza di un donatore compatibile.
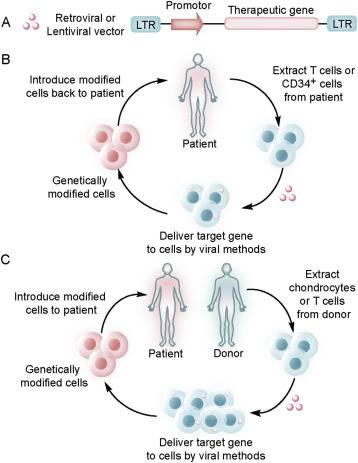
Successivamente, nel 2002, sono stadi condotti studi sperimentali in vivo sulle cellule staminali dei pazienti affetti da ADA-SCID, in cui è stato inserito, mediante un vettore di espressione, il gene ADA sano. Questi avanzamenti hanno portato nel 2016 alla commercializzazione di un farmaco, lo Strimvelis, in grado di correggere il difetto metabolico, nei casi in cui non sia possibile effettuare il trapianto di midollo. Questo ha aumentato drasticamente l’aspettativa e la qualità della vita dei pazienti affetti da deficit di adenosina deaminasi.
Fonti
- https://www.telethon.it/cosa-facciamo/ricerca/malattie-studiate/deficit-di-adenosina-deaminasi-ada-scid/
- https://www.osmosis.org/learn/Adenosine_deaminase_deficiency
- Whitmore KV, Gaspar HB. Adenosine Deaminase Deficiency – More Than Just an Immunodeficiency. Front Immunol. 2016;7:314. [PMC free article] [PubMed]
- Sauer AV, Brigida I, Carriglio N, Aiuti A. Autoimmune dysregulation and purine metabolism in adenosine deaminase deficiency. Front Immunol (2012) 3:265.10.3389/fimmu.2012.00265 [PMC free article] [PubMed] [CrossRef] [Google Scholar]
- https://www.sciencedirect.com/topics/immunology-and-microbiology/strimvelis
- https://www.microbiologiaitalia.it/immunologia/sistema-del-complemento/
Crediti immagine
- Figura 1:https://www.frontiersin.org/articles/10.3389/fimmu.2016.00314/full
- Figura 2: https://www.osmosis.org/learn/Adenosine_deaminase_deficiency
- Figura 3:https://www.sciencedirect.com/topics/immunology-and-microbiology/strimvelis