Caratteristiche ed epidemiologia
L’osteogenesi imperfetta (OI) comprende tutte quelle malattie ereditarie del tessuto connettivo che producono anomalie scheletriche con conseguente deformità e fragilità ossea. Ha un’incidenza di un caso ogni 15-20 000 nati vivi. Il primo approccio di classificazione si ha negli anni ‘80 in cui Sillence, in base alle caratteristiche cliniche e al modello di ereditarietà identifica 4 tipi di OI:
- OI ad ereditarietà autosomica dominante con sclere blu
- OI perinatale letale con femori accorciati
- OI a deformazione progressiva
- OI ad ereditarietà autosomica dominante con sclere normali
Le tipologie 1 e 4 venivano ereditate secondo un modello dominante mentre le restanti 2 e 3 secondo uno recessivo. Ciò rende visibile un fenomeno di eterogeneità genetica durante la trasmissione alla prole. La classificazione di Sillence si basava però su informazioni vincolate ad un numero ristretto di geni (soprattutto COL1A1 E COLIA2 codificanti per parti strutturali della proteina collagene).
Nel corso del tempo sono state identificate nuove mutazioni in pathway diversi, confermando così una più complessa suddivisione delle classi genotipiche e fenotipiche dell’Osteogenesi imperfetta. Adesso quindi si fa riferimento a 5 tipi diversi della malattia e le 4 precedentemente descritte da Sillence, sono utilizzate solamente per indicare il livello di gravità, in base ai parametri di frequenza di fratture e densitometria ossea secondo questo schema:
- OI di tipo1 indica lieve gravità
- OI di tipo 2 è letale
- OI di tipi 3 è severamente deformante
- OI di tipo 4 è moderatamente deformante

Eziologia e patogenesi molecolare in Osteogenesi Imperfetta
Una volta illustrate le classi diverse di OI possiamo addentrarci in quella che è la base molecolare di ognuna di esse. Ad oggi sappiamo che esistono almeno 1000 mutazioni eterozigoti dei geni maggiormente implicati quali COL1A1 E COL1A2. Esse inducono nella maggior parte dei casi Aploinsufficienza, una riduzione di circa il 50% della quantità di collagene prodotto in condizioni normali. Le mutazioni causative sono soprattutto di tipo non senso o frame-shift ma anche delezioni di un intero allele del gene COL1A1. Si evidenziano poi sostituzioni di alcune glicine con amminoacidi più piccoli tra cui cisteina, alanina o serina in estremità N-terminale del collagene.
Tra le mutazioni meno frequenti ritroviamo quelle che alterano i siti di splicing in estremità C-terminale oppure transizioni C>T nel gene che codifica per la proteina IFITM5 espressa dagli osteoblasti. Queste alterazioni appena descritte sono tipiche dell’osteogenesi imperfetta di tipo 5.
Nelle forme recessive della malattia, sono ricorrenti mutazioni omozigoti in geni che codificano per enzimi coinvolti nella biosintesi del collageno, tra cui la prolil-/lisil-ossidasi o fattori trascrizionali utili alla formazione e rimodellamento osseo.
Sintomi da alterazioni strutturali e diagnosi
In ogni caso descritto il risultato è sempre una produzione di collageno strutturalmente alterato e riconosciuto dai sistemi di controllo del nostro organismo come non-self, con conseguente messa in atto di meccanismi deputati alla sua distruzione. Per un effetto dominante negativo tutto ciò si traduce in ossa molto fragili e soggette a fratture. Ad ogni mutazione identificata corrisponde un fenotipo di una determinata gravità, ecco quindi che capiamo l’importanza della comprensione delle basi molecolari di tale patologia, sia a scopo diagnostico che prognostico.

Le indagini genetiche infatti, rappresentano lo strumento di conferma assoluto per la diagnosi di OI in supporto a quella clinica in presenza di segni caratteristici come:
- elevato numero di fratture all’anno
- deformità ossee
- sclere spesso blu scure
- faces caratteristiche ( es. viso triangolare)
Essendo però il collageno di tipo 1 ubiquitario, i segni clinici possono essere spesso generici come sordità in età precoce, alterazioni cardiache o respiratorie.
In presenza di sospetto di OI si effettua sempre uno screening del gene principalmente implicato, il COL1A1, seguita da analisi di altri papabili geni legati al processo patogenetico per individuare eventuali mutazioni secondarie meno comuni.
Un’attenta indagine genetica risulta importante non solo per la diagnosi ma anche per studiare i meccanismi di ereditarietà, il decorso e la risposta a farmaci.
Collagene: che cos’è?
Come già anticipato, le mutazioni precedentemente elencate si riflettono sulla qualità del collagene che adesso vedremo più nel dettaglio.
Si tratta della principale proteina connettivale nei mammiferi di cui il più comune sottotipo è rappresentato dal collagene di tipo 1. Esso in particolare è costituito da 3 catene a tripla elica (2 di tipo alfa-1 e una di tipo alfa-2) con andamento sinistrorso dove la sequenza amminoacidica è Gly-Pro-X oppure Gly-X OH-Pro, in cui per X si intende un amminoacido qualsiasi.
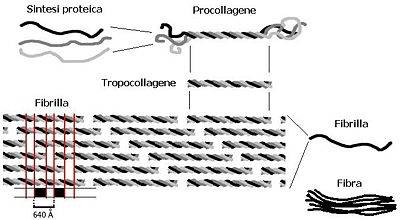
La sua biosintesi (figura 3) avviene in più step che prevedono un’iniziale trascrizione dei geni COL1A1 E COL1A2 in osteoblasti e fibroblasti dando luogo ad una forma immatura chiamata pre-pro-collagene. A seguito della perdita del peptide segnale e modifiche post traduzionali di Lys e OH-Lys diventerà tropocollagene.
Più molecole di tropocollagene si uniranno spontaneamente in fibrille che assumono una conformazione parallela e sfalsata, con una sovrapposizione tra loro per circa i ¾ della lunghezza dando luogo a fibre di collagene definitivo. Seguono ulteriori modifiche come deaminazione ossidativa di Lys e OH-Lys con lo scopo di ottenere aldeidi reattive che permettano la formazione di legami covalenti tra le varie catene per stabilizzare la struttura.
Nonostante il processo biosintetico alla base sia lo stesso, esistono diversi sottotipi di collagene:
- tipo 1 che costituisce il 90% del totale, si trova soprattutto nelle ossa, tendini, pelle e cornea ed è quello alterato nell’OI
- tipo 2 situato soprattutto in cartilagine e dischi intervertebrali
- tipo 3 presente nel sistema cardiovascolare
L’ubiquitarietà del collagene spiega la diversità dei segni clinici precedentemente elencati nelle varie tipologie di Osteogenesi imperfetta.
Test strumentali di laboratorio
Come precedentemente introdotto, il mezzo per attuare una diagnosi certa è l’analisi di geni implicati nel processo patogenetico. Essa si attua tramite un semplice prelievo di sangue per poter isolare cellule nucleate (come i leucociti) da cui poter estrarre il DNA del soggetto. Una volta estratto, viene analizzato con particolari tecniche tra cui ibridazione con sonde per identificare le più frequenti mutazioni in geni causativi come COL1A1, sequenziamento di esoni ma anche regioni introniche dei geni coinvolti con minor frequenza, sequenziamento dell’intero genoma tramite Next Generation Sequencing (NGS).
Si tratta sempre di tecniche automatizzate che riducono la probabilità di errore dell’operatore assicurando una validità totale del risultato prodotto. Vengono effettuate anche per calcolare la probabilità di avere prole affetta in caso di genitori con familiarità per Osteogenesi Imperfetta.
Terapia medica in Osteogenesi imperfetta
L’approccio medico all’OI è multidisciplinare. Si attua un’importante riabilitazione fisica mediante idro/fisioterapia con focus sulla forza muscolare e mobilità articolare per incrementare l’indipendenza dell’individuo.
Nei casi più gravi è necessario monitorare le funzionalità polmonari e cardiache poiché potrebbero essere presto compromesse da una grave scoliosi e deformità della gabbia toracica. Si ricorre anche a trattamenti ortopedici come stabilizzatori intra-midollari in ossa lunghe.
Risultano importanti anche test dell’udito già durante l’infanzia per evidenziare quei deficit uditivi neurosensoriali e conduttivi da supportare con ausili o impianti cocleari.
Nei casi più lievi invece, è sufficiente evitare quelle condizioni che possono creare traumi come sport da contatto.
Bifosfonati
Per quanto riguarda l’approccio farmacologico si somministrano i bifosfonati, degli analoghi sintetici del pirofosfato. Sono utilizzati già in età infantile, si depositano nelle superfici ossee dove si registra un’alta attività degli osteoclasti che col tempo porta ad una grave apoptosi del tessuto.
Il trattamento è volto all’aumento del volume osseo per contrastare l’alto turnover cellulare. Il nuovo osso che pian piano si forma conterrebbe così un collagene ancora difettoso ma, il volume maggiore, pareggia la scarsa qualità della proteina migliorando comunque la forza ossea.
Siccome i bifosfonati hanno un’alta emivita nell’osso (circa 10 anni), gli effetti benefici persistono a lungo anche dopo l’interruzione del trattamento. Non sono però privi di effetti collaterali.
Il loro scopo principale è quello di evitare fratture, ma l’inibizione prolungata degli osteoclasti porta ad un osso non dinamico che si traduce nella non riparazione di microfratture. Si capisce quindi che i soggetti affetti da Osteogenesi imperfetta hanno comunque un rischio maggiore di fratture ossee, soprattutto se il trattamento con bifosfonati inizia in età adulta.
Farmaci biologici: m-Ab
I progressi scientifici hanno permesso la realizzazione anche di anticorpi terapeutici con azione anabolica sulla formazione ossea. Sono ancora in via di sperimentazione tramite test su animali ma è previsto a breve l’inizio di studi pediatrici. In particolare troviamo la sclerotina, un regolatore negativo della formazione ossea nel WNT pathway e il fattore di crescita trasformante β, un coordinatore del rimodellamento osseo degli osteoblasti. Nel primo caso l’anticorpo aumenta la massa ossea senza inibire il turnover cellulare mentre nel secondo si va anche a ridurre il ricambio osseo.
Prospettive future
L’Osteogenesi imperfetta rimane una patologia dalla prognosi negativa poiché attualmente non esistono farmaci in grado di arrestare il processo patogenetico alla base. E’ una malattia fortemente invalidante che può causare la morte durante la vita intra-uterina. In casi più lievi permette una vita abbastanza normale in cui però non deve mai mancare l’attenzione verso quelle azioni quotidiane che potrebbero aumentare notevolmente il rischio di fratture.
E’ stato lungo il cammino verso la comprensione dei modelli di ereditarietà poiché fortemente atipici ma, grazie alle nuove tecnologie, sono stati raggiunti comunque traguardi importanti.
Sicuramente un ruolo chiave è rappresentato dal sequenziamento di nuova generazione (NGS) rivolto all’esoma e metabolomica ossea per la conoscenza di ulteriori mutazioni da impiegare sia a scopo diagnostico che terapeutico.
Fonti
- https://pubmed.ncbi.nlm.nih.gov/26542481/
- https://pubmed.ncbi.nlm.nih.gov/24715559/
- Connective tissue and its heritable disorders: molecular, genetic and medical aspects 2nd edition, Peter M. Royce, Beat Steinmann
- https://pubmed.ncbi.nlm.nih.gov/21829228/
- https://pubmed.ncbi.nlm.nih.gov/23602968/
- https://pubmed.ncbi.nlm.nih.gov/20592128/
Crediti immagini:
- immagine in evidenza: https://www.osservatoriomalattierare.it/malattie-rare/osteogenesi-imperfetta/14690-osteogenesi-imperfetta-la-storia-di-chiara-performer-che-dona-il-suo-corpo-a-servizio-della-scena
- Figura 1: storiadellamedicina.net
- Figura 2: i malati invisibili
- Figura 3: Wikipedia