Definizione
Nel 1965, il medico inglese Harry Angelman descrisse 3 pazienti che presentavano un’andatura rigida, a scatti, assenza di linguaggio, risate eccessive e convulsioni. Questo disturbo prese il nome di sindrome di Angelman e colpisce circa 1 persona su 15.000. La Sindrome di Angelman (Fig.1) è una malattia genetica che interessa in modo specifico il sistema nervoso centrale e lo sviluppo neuro-comportamentale. Il ritardo dello sviluppo negli individui con la Sindrome di Angelman si osserva di solito entro il primo anno di vita. A causa di un complesso meccanismo biologico, che verrà approfondito in seguito, chiamato imprinting, i geni contenuti in questa porzione del cromosoma 15 sono funzionanti solo nel cromosoma materno, e sono “spenti “in quello paterno. Di conseguenza i disturbi del movimento includono tremori, scatti e atassia.
Eziologia e Patogenesi
È causata da diversi meccanismi genetici che determinano un’alterata funzione o regolazione della copia del gene UBE3A ereditata dalla madre. Il gene è localizzato sul braccio lungo del cromosoma 15, nella regione q11.2-q13. Ogni essere umano eredita una copia del gene UBE3A da ciascun genitore ed entrambe le copie vengono utilizzate nei vari distretti corporei, ma in alcune specifiche aree del cervello è la sola copia materna ad essere correttamente attivata. Quando la copia materna del gene UBE3A viene persa/modificata a causa di un cambiamento cromosomico o di una mutazione genetica, l’individuo perderà quindi la capacità di attivazione del gene nei distretti dove la funzionalità è limitata alla copia della madre. Detto questo, le anomalie genetiche che determinano la Sindrome di Angelman sono:
- delezioni materne de novo del cromosoma 15q11-q13 nel 70–80 % dei casi
- mutazioni intrageniche nel cromosoma UBE3A ereditato dalla madre
- disomia uniparentale paterna per il cromosoma 15q11-q13 nel 3–5 % dei casi (UPD)
- difetti di imprinting all’interno del cromosoma 15q11-q13 che alterano l’espressione di ereditarietà materna (IC)
Pertanto, i test genetici molecolari (analisi della metilazione e analisi della sequenza UBE3A) identificano alterazioni in circa il 90% degli individui. Il restante 10% degli individui con caratteristiche fenotipiche classiche della Sindrome di Angelman presentano un meccanismo genetico non ancora identificato.
Sintomatologia
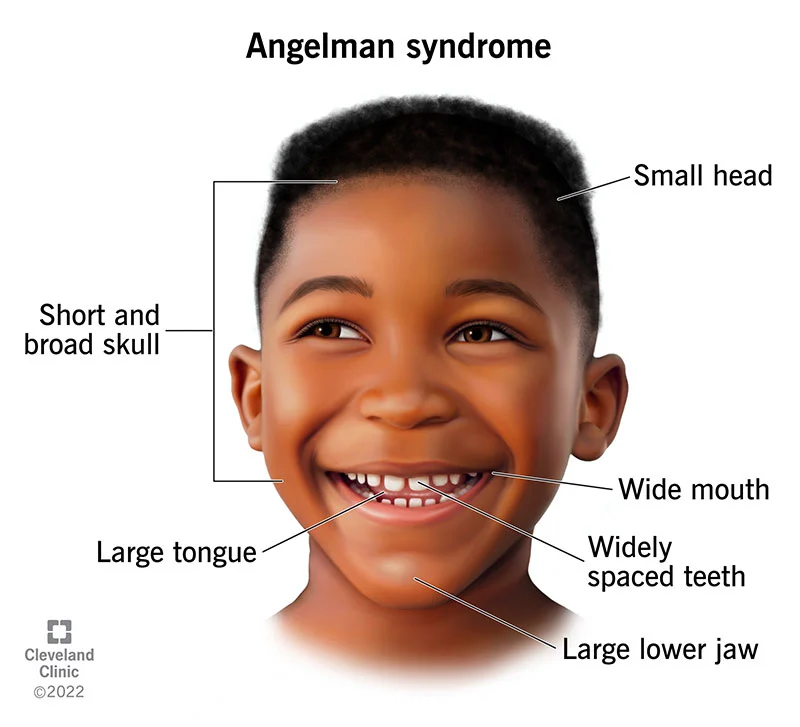
I soggetti affetti dalla Sindrome di Angelman possiedono sia caratteristiche fisiche particolari, sia un deficit cognitivo, che determina molti sintomi e problematiche. Dal punto di vista dei tratti somatici (Fig.2) essi presentano:
- cranio corto e largo
- lingua larga
- testa piccola
- bocca grande
- denti ampiamente distanziati
- grande mascella inferiore
- carnagione chiara
Mentre da un punto di vista cognitivo:
- ritardo mentale grave
- mancanza di parola, ma alcuni soggetti che sono leggermente colpiti possono acquisire poche parole
- iperattività
- risata facile
- attrazione per l’acqua
- riduzione del sonno
Diagnosi
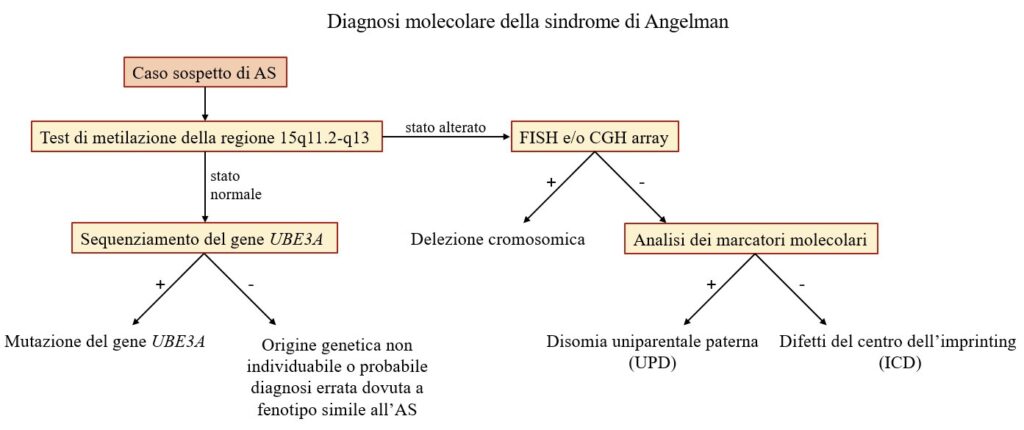
La diagnosi della sindrome di Angelman non comprende solamente l’aspetto fisico ma anche un’analisi genetica. Sebbene nella maggior parte dei casi la diagnosi è clinica, la conferma molecolare è fondamentale per evidenziare il tipo specifico di alterazione molecolare perché offre importanti elementi aggiuntivi. Il primo è la possibilità di individuare il possibile rischio di ricorrenza nella famiglia. In modo particolare, la causa più frequente (70%) è la microdelezione della regione 15q11.2-q13 di derivazione materna, mentre molto più rara è la disomia uniparentale paterna (UPD).
In altri casi (2-3%), la sindrome è dovuta a difetti primari nel centro dell’imprinting (ICD) con perdita di metilazione del DNA. Mutazioni sull’allele materno del gene UBE3A, infine, sono causative della malattia nel 5-7% dei pazienti. Per evidenziare la lesione genetica della sindrome di Angelman è necessario procedere con più approcci molecolari, proprio per cercare di evidenziare l’alterazione nel singolo paziente. Nella Fig. 3 è indicato uno schema del percorso diagnostico.
L’analisi della sequenza UBE3A rileva invece varianti patogenetiche in un ulteriore 11% circa degli individui che causano la assenza della proteina codificata da UBE3A o la sua scarsa funzione.
Test utilizzati
I test che vengono maggiormente utilizzati sono:
- approccio citogenetico: permette di evidenziare cambiamenti dell’assetto cromosomico, come delezioni, duplicazioni o riarrangiamenti strutturali. La più comune delezione di 5-7 Mb (mega basi di sequenza) non è generalmente rilevata dall’analisi cromosomica di routine.
- test di metilazione della regione 15q11.2-q13, eseguito mediante MS-PCR (Methylation Specific PCR), pirosequenziamento e, più recentemente, mediante MS-MLPA (Methylation Specific Multiplex Ligation – Dependent Probe Assay), permette di rilevare microdelezioni, disomie uniparentali (UPD) o difetti nel centro dell’imprinting (ICD), confermando la diagnosi in circa l’80% dei pazienti
- Il test di ibridazione fluorescente in situ (FISH), rileva la presenza di una delezione di circa 4-6 Mb nella regione 15q11.2-q13, presente in circa il 70% dei pazienti AS (Fig. 4). Il microarray cromosomico (CMA) può ulteriormente affinare la dimensione della delezione, che è stata dimostrato essere correlata alla gravità del fenotipo clinico. Pertanto se un individuo risulta positivo sia al test di metilazione sia all’analisi FISH, è possibile confermare che il soggetto presenta la Sindrome di Angelman causata da delezione. Se un individuo risulta, invece, positivo al test di metilazione, ma il test FISH è negativo, avrà probabilmente UPD o ICD. L’analisi di marcatori molecolari verrà, quindi, utilizzata per determinare se l’individuo ha due copie del cromosoma 15 del padre (UPD) o se l’individuo ha un cromosoma 15 ereditato da ciascun genitore, ma con metilazione errata (ICD). In quest’ultimo caso, anche se l’individuo ha ereditato una copia del cromosoma 15 dalla madre e, quindi, il gene UBE3A è presente, il pattern di metilazione stabilito non è corretto e il gene di origine materna è silenziato.
Alle famiglie con un caso di sindrome di Angelman dovrebbe essere offerta una consulenza genetica poiché mutazioni del gene UBE3A possono comportare un rischio di ricorrenza fino al 50%.

Terapie
La terapia consiste in una combinazione variabile di supporti specialistici, calibrati in base alle caratteristiche peculiari di ogni paziente. Per questo motivo è indicato eseguire esami ematici di routine. Tra le terapie che possono rientrare nel trattamento di un paziente affetto da Sindrome di Angelman ci sono:
- farmaci antiepilettici
- fisioterapia, per il trattamento delle difficoltà di movimento, coordinazione ed equilibrio
- logopedia ed altre terapie volte a favorire la comunicazione,
- terapia comportamentale per gestire iperattività e ridotta capacità di attenzione
- monitoraggio oculistico
La loro aspettativa di vita è normale. Sebbene questa sia la situazione attuale, il futuro riserva però grandi speranze di cura. Gli scienziati credono infatti che la Sindrome di Angelman abbia il più grande potenziale di cura tra tutte le malattie neurogenetiche.
Fonti:
- MARGOLIS, Seth S., et al. Angelman syndrome. Neurotherapeutics, 2015, 12.3: 641-650.
- https://www.ospedalebambinogesu.it/sindrome-di-angelman-80362/
- https://healthy.thewom.it/salute/sindrome-angelman/
- WILLIAMS, Charles A. Neurological aspects of the Angelman syndrome. Brain and Development, 2005, 27.2: 88-94.
- https://www.osservatoriomalattierare.it/malattie-rare/sindrome-di-angelman
- https://www.cureangelman.it/conosci-la-sindrome-di-angelman/
Crediti immagini:
- Immagine in evidenza: http://www.medicocontesta.com/2017/01/el-sindrome-de-angelman.html
- Figura 1: https://www.france-assos-sante.org/2020/01/15/temoignages-parents-enfants-angelman/
- Figura 2: https://my.clevelandclinic.org/health/diseases/17978-angelman-syndrome
- Figura 3: https://www.cureangelman.it/conosci-la-sindrome-di-angelman/
- Figura 4: https://www.cureangelman.it/conosci-la-sindrome-di-angelman/