Caratteristiche
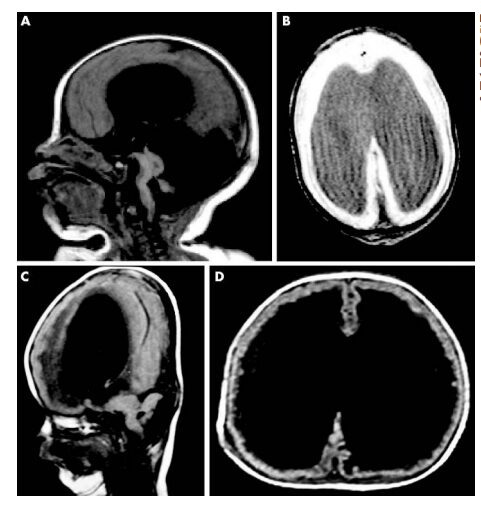
La Sindrome di Walker-Warburg (WWS) è una rara forma di distrofia muscolare, nonché patologia genetica autosomica recessiva, associata allo sviluppo di malformazioni oculari e cerebrali (Fig.1). La patologia è identificata nel 10-20% dei casi tramite analisi genetiche volte a ricercare mutazioni sul gene della proteina O-mannosiotransferasi 1 (POMT1), e sui geni adiacenti (per conferma) fukutina, FKRP (Proteina Correlata alla Fukutina), e POMT2.
La sua distribuzione è globale, e la sua incidenza nel Nord-Est dell’Italia nei primi anni 2000 è stata di 1,2 casi su 100.000 nascite.
Le analisi di laboratorio evidenziano nei pazienti un’aumentata produzione di creatin-chinasi e di alfa-distroglicano alterato, inoltre, le analisi prenatali con ultrasuoni possono evidenziare problematiche anche in famiglie la cui storia genetica non fosse nota.

{kind=link}
Eziologia
Il Complesso della Glicoproteina Distrofina (DGC; Fig.2) è composto da un insieme di proteine collocate a livello del sarcolemma delle fibrocellule muscolari.
Difetti a carico di questo complesso sono spesso associati alle distrofie muscolari provocando, come conseguenza, problemi nell’organizzazione della membrana basale.
Il distroglicano, componente essenziale del DGC, è espresso in numerose cellule sotto forma di precursore propeptidico che viene successivamente clivato e variamente glicosilato per dare l’alfa– e il beta-distroglicano attivi.
Secondo studi recenti, sarebbe un particolare tipo di tetrasaccaride dell’alfa-distroglicano a rappresentare il punto di ancoraggio alla laminina e ad altri membri della membrana basale. Perciò, mutazioni a carico di questo tetrasaccaride che impediscono il corretto legame del distroglicano all’ambiente extracellulare, riducono la forza di ancoraggio delle fibre muscolari tra di loro, con conseguente disfunzione muscolare e necrosi delle fibre.
Nei pazienti affetti da distrofie muscolari, infatti, l’alfa-distroglicano è scarsamente evidenziato da tecniche immuno-enzimatiche poichè scarsamente glicosilato.

Analisi di linkage genome-wide in 10 famiglie con storia correlata alla Sindrome WW, hanno evidenziato l’esistenza di 3 loci genici associabili alla patologia.
Mutazioni a carico della proteina O-mannoside beta-1,2-N-acetilglucosamiltransferasi (POMGnT1) sono emerse come responsabili delle anomali oculari.
Inoltre, mutazioni omozigoti (5) nel locus del gene POMT1 sul cromosoma 9q34 sono state identificate in cinque dei pazienti consanguinei analizzati.
Tra le 5 mutazioni in questione, due erano non senso, due di scorrimento della finestra di lettura (frameshift) e una missenso.
Quando le mutazioni si presentano in forma omozigote nei pazienti, determinano la comparsa di un fenotipo più severo della patologia, al contrario, quando sono presenti in eterozigosi risultano essere associate ad un fenotipo più blando e ad un’aspettativa di vita maggiore.
Altri casi di mutazione
In un altro soggetto analizzato è stata identificata una mutazione omozigote per il gene della fukutina, e in un altro ancora una mutazione missenso per il gene FKRP.
Nel 2005 è stato poi identificato un altro gene potenzialmente responsabile della patologia, ovvero, POMT2 con mutazioni omologhe riscontrate in tre famiglie diverse.
Il genoma umano contiene due geni codificanti per POMPT (POMT1 e POMT2), in tutti i tessuti il primo codifica per un mRNA di 3.1 kb con livelli particolarmente elevati nei testicoli e nel cervello a livello fetale. E’ lo splicing alternativo su diversi esoni a determinare quale sarà l’isoforma finale della proteina in questione.
I due geni devono essere co-espressi correttamente per ottenere un giusto livello di enzima POMT.
Patogenesi
Nei tessuti cerebrali dei topi, la delezione selettiva del distroglicano è sufficiente per portare allo sviluppo di malformazioni al cervello con disallineamento delle cellule corticali, fusione degli emisferi con folia cerebellare e aberrante migrazione delle cellule granulari.
Tutto questo è dovuto ad uno scarso ancoraggio alla laminina della membrana basale, essenziale per la corretta migrazione cellulare.
Le analisi immunoistochimiche e quelle al microscopio elettronico condotte sui tessuti muscolari di pazienti con Sindrome di Walker-Warburg, hanno evidenziato la presenza di mutazioni omozigoti di frameshift sul gene POMT1. La catena alfa-2 della laminina e del perlecano erano presenti nelle fibre muscolari e nei nervi intramuscolari periferici; assente l’epitopo alfa-distroglicano glicosilato.
Inoltre, la membrana basale delle fibre muscolari appariva spesso distaccata e non in continuità completa con il plasmalemma cellulare.
Diverse tipologie di nuclei, come i mionuclei e i nuclei satellite, erano completamente privi di eterocromatina a livello della membrana nucleare; cambiamenti apoptotici erano presenti nel 3% delle fibre muscolari.
Questi meccanismi errati e queste mancanze sono, secondo i più recenti studi, le cause da impure allo sviluppo di questa complessa patologia associata a processi degenerativi a carico muscolare e cerebrale.
Segni e sintomi
La Sindrome di Walker-Warburg (WWS) provoca distrofia muscolare, lissencefalia di II tipo, idrocefalo, e anomalie cerebrali e oculari (Fig.3).
Si tratta di una delle tipologie di distrofie muscolari più severe, e i suoi segni e sintomi sono già chiaramente visibili fin dall’infanzia.
I pazienti presentano una generale ipotonia, debolezza muscolare, ritardo mentale e talvolta epilessia.
Molte sono le anomalie oculari come: cataratte, microcornea, microftalmia, difetti del cristallino, displasia retinica, coloboma e glaucoma.
Per quanto riguarda le anomalie cerebrali, queste riguardano: lissencefalia di tipo II con corteccia cerebrale totalmente disfunzionale, ipoplasia cerebellare e danni al tronco encefalico.
Inoltre, possono essere presenti ulteriori problematiche come l’ipoplasia del corpo calloso, encefalocele occipitale e malformazioni tipicamente associate alla Sindrome di Dandy-Walker.
Altri sintomi possono comprendere un pene di piccole dimensioni con testicoli ritenuti, orecchie prominenti e cheiloschisi del palato.
Epidemiologia
La distruzione della Sindrome di Walker-Warburg è globale, tuttavia, non esiste un dato di incidenza completo a livello planetario.
Come esposto in precedenza, nei primi anni 2000 nel Nord-Est del nostro paese, l’incidenza era pari a 1,2 casi su 100.000 nati.
Diagnosi e test di laboratorio
La diagnosi della Sindrome WW si basa su diversi aspetti:
- sulla ricerca di distrofie muscolari congenite associate alla ipoglicosilazione dell’alfa-distroglicano;
- su alti livelli di creatin-chinasi;
- anomalie anteriori e posteriori degli occhi;
- difetti nella migrazione neuronale con lissencefalia di II tipo e idrocefalo;
- anomalie cerebellari.
Le mutazioni o i locus genici associati alla patologia sono ad oggi piuttosto ignoti.
Solo nel 10-20% dei casi, infatti, si riscontrano mutazioni per i geni POMT1, POMT2, o FKRP.
Una diagnosi prenatale è possibile in famiglie in cui è nota la presenza delle mutazioni, inoltre, gli ultrasuoni possono mettere facilmente in evidenza la presenza delle anomalie associate.
L’idrocefalo, per esempio, può essere osservato già durante la XIII settimana di gestazione, mentre l’encefalocele intorno alla XVIII settimana.
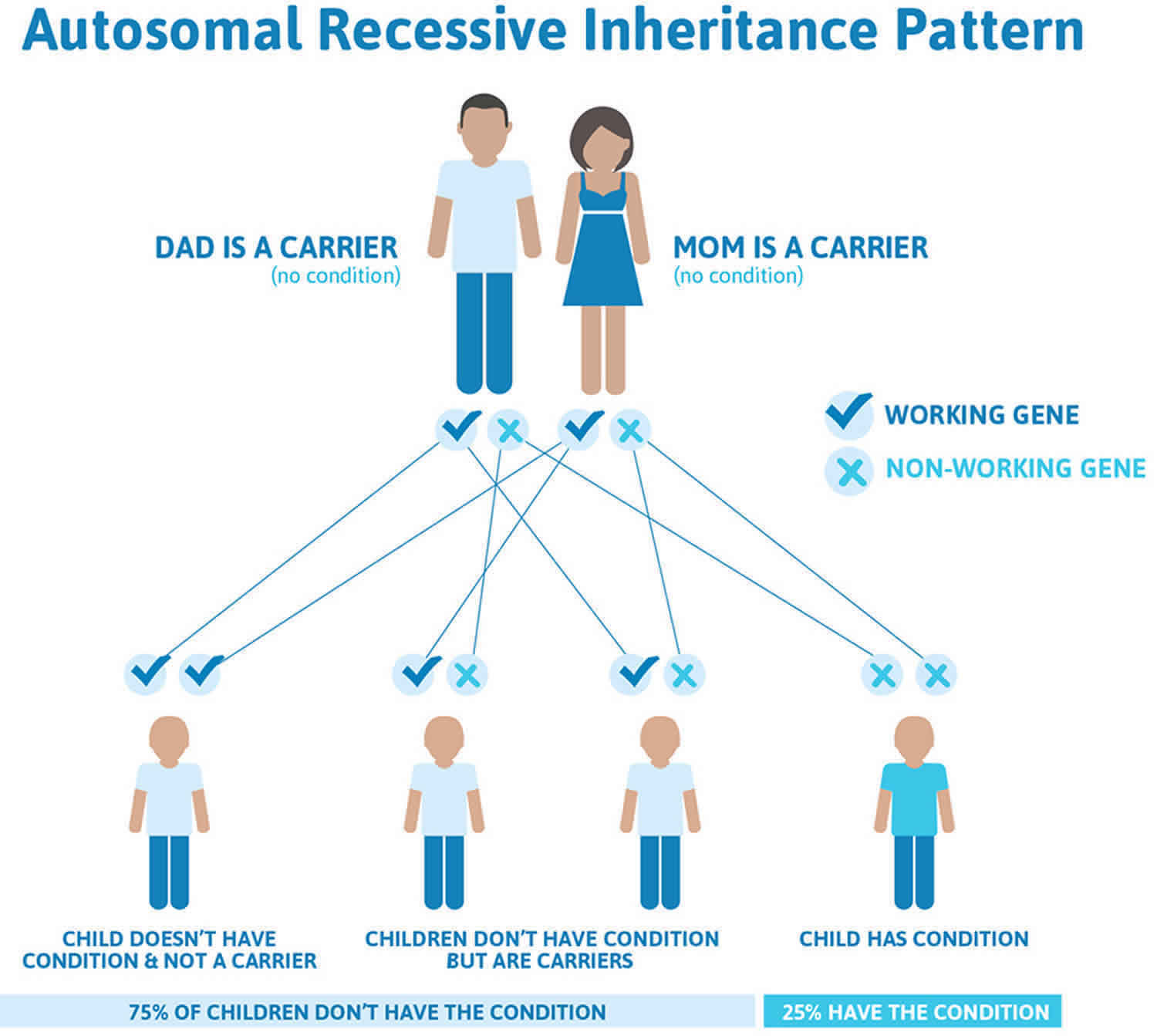
Si tratta di una patologia autosomica recessiva che in famiglie presentanti già un figlio malato porta ad una probabilità del 25% di averne un altro affetto (Fig. 4).
{kind=link}
Terapia
Le cure sono solamente di supporto e preventive.
Per esempio, se compaiono i primi sintomi di crisi epilettiche è possibile intervenire con farmaci anticonvulsivanti, inoltre, numerose pratiche chirurgiche possono alleviare i sintomi legati all’idrocele e all’encefalocele.
Sono quindi possibili diversi trattamenti terapici volti, non a curare la patologia, ma a facilitare la vita dei pazienti affetti.
Fonti
- Vajsar J., Schachter H. (2006). Walker-Warburg syndrome. Orphanet Journal of Rare Disease 1:29, pp.1-5.
- William B. Dobyns M.D., Roberta A. Pagon, D. Armstrong et al. (1989). Diagnostic criteria for Walker-Warburg syndrome. American Journal of Medical Genetics 32(2), pp.195-210.
- Mustacciuolo M.L., Miorin M., Martinello M. et al. (1996). Genetic epidemiology of congenital muscular dystrophy in a sample from the north-east Italy. Human Genet 97, pp. 277-279.
- Rarediseases.info.nih.gov
- National library of medicine
Crediti immagini
- Immagine in evidenza: sociedadcanariadeoftalmologia.com;
- Figura 1: ojoonline.org;
{kind=link}
non sappiamo nulla su eventuali ipotesi di eziologia ambientale ? famiglie povere, epsosizioni occupazionali dei genitori, gruppo etnico ?
Grazie
Vito Totire