Cosa sono le proteine ? Quali funzioni assumo in base al loro “folding”? Come fa il software AlphaFold a “predirne” la struttura ?
Le proteine sono molecole biologiche fondamentali per il nostro organismo. Hanno varie forme e funzioni, fra cui:
- Strutturale
- Immunitaria
- Trasporto
- Ormonale
- Energetica
- Enzimatica_ cioè accelerano le reazioni chimiche nel nostro corpo, rendendole “a tempo di vita”, es. la lattasi scinde il lattosio (zucchero del latte) in glucosio e galattosio.
Non ci stupisce quindi che esistono migliaia di proteine, con forme e proprietà chimiche diverse fra loro, in aree specifiche della cellula.
Tutta questa diversità può esser ricondotta ad una sequenza lineare di aminoacidi, analoga ad una “collana di perle”. Proprio sulla sequenza e sulle conformazioni che essa assume, che lavora il software AlphaFold; promuovendo lo studio strutturale della proteina.
Gli amminoacidi che compongono le proteine, sono sempre gli stessi 20, ma in varie configurazioni e disposizioni nelle differenti proteine. Queste sequenze si andranno poi a ripiegare su se stesse, a formare una struttura tridimensionale con particolari proprietà chimico/fisiche.
Le proprietà delle proteine, dipendono dalla natura degli aminoacidi di cui sono composte.
Tipologia di amminoacidi che troviamo nelle proteine:
- ingombranti, che non permettono il ripiegamento in alcuni punti della struttura proteica
- non ingombranti, lasciano più gradi di liberta al ripiegamento.
- carichi positivamente e negativamente.
- idrofilici (che essendo affini all’acqua vengono esposti all’esterno della proteina).
- idrofobici (che non sono affini all’acqua e si dispongono a l’interno della proteina o fra i lipidi che compongono la membrana cellulare).
Quindi in teoria conoscendo la sequenza di aminoacidi, si dovrebbe sapere come “folda” la proteina; ma questo è praticamente impossibile, dato che i vari monomeri che compongono la struttura proteica, sono sottoposti a moto browniano e quindi esistono infinite combinazioni che essi possono assumere nello spazio. Questa problematica ha rappresentato un grande scoglio nello studio del “folding proteico” negli ultimi 50 anni.
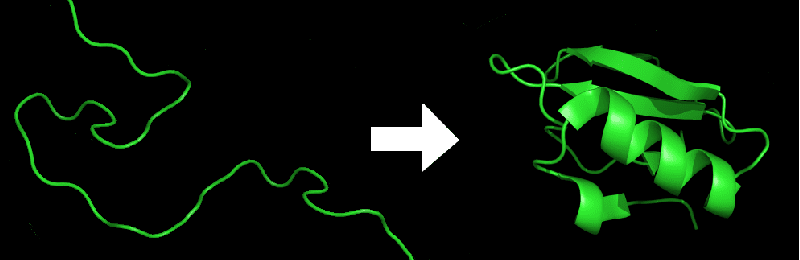
In realtà dalla struttura primaria delle proteine, si può risalire a grandi linee al “folding finale”, ma sapere con esattezza come ogni amminoacido si dispone nella struttura è impossibile: o meglio era impossibile ! Infatti oggi grazie al software AlphaFold che “predice” attraverso algoritmi la struttura delle proteine, si possono determinare con facilità i vari ripiegamenti che il polimero assume.
Come si determina la struttura di una proteina
Ad oggi una delle tecniche più usate per determinare la struttura di una proteina è la cristallografia, un lavoro molto certosino che è stato usato per decenni ed ha permesso di ricavare circa 190.000 strutture.
Il dato fornito in precedenza corrisponde però a circa un millesimo di tutte le proteine di cui abbiamo la sequenza.
Prima di studiare la proteina bisogna infatti, sequenziare il gene che la esprime.
Capiamo subito che è molto più facile sequenziare un gene, grazie alle nuove tecniche di sequenziamento molto avanzate, che studiarne la struttura fine del suo prodotto.
La corsa al folding proteico: il ruolo di DeepMind nello sviluppo del software AlphaFold applicato studio delle proteine
Per decenni biologi, fisici, chimici e bioinformatici hanno cercato di predire con dei software il folding delle proteina ma con risultati molto deprimenti, fino a quando non è entrata in gioco DeepMind.
Si tratta di un’azienda acquistata da Google nel 2014 ed è una realtà che si occupa di intelligenza artificiale.
DeepMind è un azienda pionieristica nell’ambito delle di tecniche di deep learning (cioè si prende una rete neurale, e gli si danno “in pasto” dei grandi data set, sui quali il software si allena, in base agli obbiettivi impostati; ad esempio riconoscere una proteina).
DeepMind era già famosa per alcune intelligenze artificiali nell’ambito di giochi da tavolo come gli scacchi e Go, dove era anche riuscita a sconfiggere i campioni mondiali.
Dopo essere stata acquistata da Google DeepMind, ha iniziato lo sviluppo di una nuova intelligenza artificiale, AlphaFold, che ha l’obbiettivo di conoscere la struttura tridimensionale delle proteine a partire delle sequenze amminoacidiche ottenibili da data set.
I primi risultati ed ascesa del software AlphaFold nello studio delle proteine
Nel 2020 i primi risultati dell’intelligenza artificiale erano già strabilianti, battendo tutti gli altri programmi che funzionavano in modo diverso, specialmente nei confronti del programma RoseTTAFold, suo principale competitor.
Nel 2021, AlphaFold, rilascia 1.000.000 di strutture e quindi in poco più di un anno dalla prima pubblicazione vengono rese note ben 5 volte il numero delle strutture delle proteine che fino ad allora l’uomo era riuscito a ottenere con metodi classici.

Questo è molto utile a livello di ricerca ad esempio si può conoscere il livello di conservazione di una proteina nelle varie specie e quindi utilizzarla per eventuali comparazioni filogenetiche.
La cosa ancora più interessante ci arriva a fine luglio 2022, quando DeepMind ha dichiarato di aver predetto oltre le 200.000.000 di strutture proteiche. Tali dati sono presenti online sul sito AlphaFold e nelle varie banche dati come Uniprot.
Potenziali aggiuntivi di AlphaFold nello studio del “fold” proteico
AlphaFold fornisce anche il livello di confidenza fra le varie aree della proteina, quindi ci indica la probabilità che l’intelligenza artificiale ha di rappresentare il reale fold proteico.
Questa potenzialità del programma è molto interessante dato che si attribuisce la probabilità che quella conformazione proteica appartenga realmente alla realtà.
La confidenza si esprime in pLDDT e corrisponde al punteggio previsto del modello sulla metrica lDDT-Cα, con una scala da 0 a 100:
- Le regioni con pLDDT > 90 dovrebbero essere modellate con elevata precisione.
- Le regioni con pLDDT tra 70 e 90, hanno una previsione di backbone generalmente buona.
- Le regioni con pLDDT tra 50 e 70 sono a bassa confidenza.
- Le coordinate 3D delle regioni con pLDDT <50 hanno spesso un aspetto simile a un nastro e non devono essere interpretate, infatti sono un fattore di disturbo.
Limiti di AlphaFold
- Non genera previsioni multi-catena (complessi).
- Non è stato convalidato per prevedere l’effetto delle mutazioni.
- Non prevede le posizioni dei componenti non proteici nelle strutture sperimentali (come cofattori, metalli, ligandi, ioni, DNA/RNA o modifiche post-traduzionali).
Conclusioni
Dai risultati ottenuti dall’intelligenza artificiale compaiono anche strutture di proteine mai viste fin ora.
Esplorare il sito AlphaFold può essere una scoperta continua in ogni momento, dato che possiamo imbattertici in strutture proteiche che fino ad allora erano sconosciute a l’umanità.
Quindi da ora in poi si cambierà il modo di studiare le proteine, dato che l’intelligenza artificiale affiancherà sempre di più i metodi classici di studio cristallografici, svolgendo un lavoro di ricerca molto “più fine”, con conseguente maggior comprensione dei fenomeni biologici.
Fonti:
Immagini:
- Figura 1:https://aulascienze.scuola.zanichelli.it/multimedia-scienze/science-news/ripiegamento-delle-proteine-in-diretta
- Figura 2:https://www.deepmind.com/