Prioni: cosa sono?
Il termine prione è stato utilizzato per la prima volta nel 1982 dal biochimico e neurologo Stanley B. Prusiner per identificare una sorta di agente infettivo proteico in grado di causare diverse patologie neurodegenerative. L’ipotesi dell’esistenza di proteine come particelle infettive fu inizialmente abbastanza sconvolgente, in quanto non erano noti agenti infettivi privi di materiale genetico, ma questa spiegazione sembrava l’unica valida ed adeguata per descrivere come queste particelle fossero resistenti ai raggi UV (che causano danni agli acidi nucleici), ma allo stesso tempo suscettibili a proteasi.
Ancora più sconvolgente fu accorgersi di come queste proteine fossero per la maggior parte presenti anche nelle cellule normali. Quello che viene chiamato prione ne è infatti una variante che differisce solamente per un’alterazione nella conformazione. Un’ipotesi successiva speculò inoltre che il meccanismo infettivo di propagazione potesse consistere in un legame tra la proteina sana e la stessa proteina alterata (prione) in grado di convertire la proteina sana nella conformazione alterata.
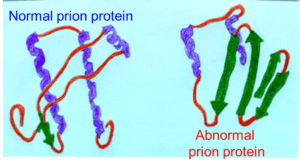
PrP
La proteina che si crede responsabile delle malattie da prioni è stata chiamata PrP (figura 1); nella sua forma fisiologica (PrPc), essa è presente in ognuno di noi e non causa patologia. Questa proteina è composta da 253 amminoacidi, è codificata da un gene situato nel braccio corto (p) del cromosoma 20 e viene espressa in maniera particolarmente abbondante a livello cerebrale (figura 2).

Le malattie prioniche
Le malattie da prioni sono state associate a forme infettive, ereditarie, ma anche a rari casi sporadici. Il meccanismo alla base sembra comunque essere lo stesso, ovvero la presenza di PrP mutata per qualche motivo (gene alterato nel caso delle ereditarie, inoculazione nel caso delle infettive o mutazione nel caso delle sporadiche).
Il processo che porta alla mutazione di conformazione non è compreso del tutto, così come neanche il ruolo fisiologico della proteina normale. Prpc è ancorata alla membrana cellulare tramite il GPI (Glicosil-fosfatidil-inositolo) e può legare un atomo di rame nella sua porzione ammino-terminale. Per queste ragioni si ritiene possa avere un ruolo nella trasduzione del segnale o nella protezione da stress ossidativo, anche se le evidenze sono ancora carenti.
Prioni e mucca pazza
La conoscenza su questo tipo di agente infettivo è aumentata esponenzialmente da quando si sono verificati diversi casi di BSE (encefalopatia spongiforme bovina), la cosiddetta malattia della mucca pazza, negli anni ’90; ma siamo ancora lontani dalla comprensione esaustiva di questo meccanismo e, di conseguenza, da una cura per tale patologia.
Bibliografia ed approfondimenti
- https://en.wikipedia.org/wiki/PRNP
- http://www.treccani.it/enciclopedia/prione/
- https://www.scientificamerican.com/article/what-is-a-prion-specifica/
Credits foto
- https://en.wikipedia.org/wiki/PRNP
- https://phys.org/news/2009-02-sticky-antibodies-block-prion-disease.html (immagine in evidenza, anticorpi legati a proteine prioniche).
I commenti sono chiusi.