Introduzione
La tau è una proteina legante i microtubuli e si ritiene svolga un ruolo importante nell’assemblaggio e nella stabilizzazione degli stessi. I microtubuli, a loro volta, sono fondamentali per il trasporto intracellulare e per il mantenimento della forma e della funzione neuronale.
Isoforme della proteina tau
Nel cervello umano, esistono sei isoforme della proteina Tau prodotte a partire dal gene MAPT (Microtubule-Associated Protein Tau) attraverso un meccanismo di splicing alternativo dell’mRNA.
{kind=link}
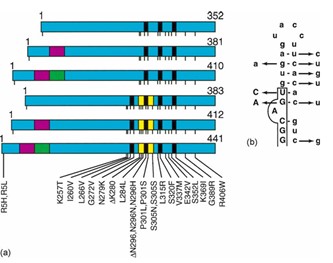
Tali isoforme differiscono tra di loro per la presenza/assenza di un inserto di 29 o di 58 aminoacidi all’estremità ammino-terminale della proteina e per l’inclusione o meno, di una sequenza di 31 aminoacidi ripetuti (codificata dall’esone 10 della proteina tau) in corrispondenza dell’estremità carbossi-terminale. L’esclusione dell’esone 10 porta alla produzione di tre isoforme, ciascuna contenente tre ripetizioni. Al contrario, la sua inclusione, porta alla formazione di altre tre isoforme ciascuna contenente quattro ripetizioni (Figura 1).
Le mutazioni tau sono:
- mutazioni missenso;
- delezioni;
- mutazioni silenti nella regione codificante;
- mutazioni introniche localizzate vicino al sito donatore di splicing dell’introne che segue l’esone 10 sottoposto a splicing alternativo (Fig. 1).
Funzionalmente, rientrano in due categorie in gran parte non sovrapposte: quelle il cui effetto primario è a livello proteico e quelle che influenzano lo splicing alternativo del pre-mRNA di tau. La maggior parte delle mutazioni missenso riduce la capacità della proteina tau di interagire con i microtubuli, in particolare di promuoverne l’assemblaggio.
Malfunzionamento della proteina tau associata a patologie neurodegenerative
L’accumulo della proteina tau è strettamente associato a diverse malattie neurodegenerative, note collettivamente come taupatie. Queste includono l’Alzheimer, la demenza frontotemporale e la paralisi sopranucleare progressiva. La proteina tau, normalmente solubile e funzionale, diventa patologica quando si iperfosforila e si aggrega in filamenti elicoidali e fibrille.
L’iperfosforilazione della tau è un evento critico che precede la sua aggregazione. Questo processo altera la conformazione della proteina, portando alla formazione di strutture insolubili che si accumulano nei neuroni. Questi aggregati interferiscono con la funzione cellulare, contribuendo alla neurodegenerazione. Le taupatie sono caratterizzate da una progressiva perdita neuronale e da disfunzioni sinaptiche, che si traducono in deficit cognitivi e motori.
La fosforilazione anomala della tau è mediata da diverse proteine, tra cui la GSK-3β (Glycogen Synthase Kinase 3 beta) e la CDK5 (Cyclin-Dependent Kinase 5). Queste chinasi aggiungono gruppi fosfato a specifici residui amminoacidici della tau, inducendo cambiamenti conformazionali che promuovono l’aggregazione.
Una volta iperfosforilata, la tau perde la sua capacità di legarsi ai microtubuli e inizia a formare oligomeri solubili. Questi oligomeri possono ulteriormente aggregarsi in strutture più grandi, come i filamenti elicoidali e le fibrille. Gli aggregati di tau possono così interagire con altre proteine cellulari, “disturbando” vari processi cellulari e contribuendo alla neurodegenerazione.
Fattori genetici ed ambientali coinvolti nell’accumulo della proteina tau
I fattori genetici giocano un ruolo significativo nell’accumulo delle proteine tau. Mutazioni nel gene MAPT sono state associate a diverse taupatie ereditarie. Queste mutazioni possono influenzare la struttura e la funzione della tau, aumentando la sua tendenza all’aggregazione. Oltre ai fattori genetici, anche quelli ambientali possono contribuire all’accumulo degli oligomeri. Stress ossidativo, infiammazione cronica e traumi cranici sono stati identificati come potenziali fattori di rischio in grado di indurre modifiche post-traduzionali delle tau promuovendone, quindi, l’aggregazione.
Ruolo protettivo delle Tau contro lo stress ossidativo
Ricercatori del Baylor College of Medicine e del Jan and Dan Duncan Neurological Research Institute (Duncan NRI) del Texas Children’s Hospital hanno scoperto un altro ruolo positivo della proteina tau. Essa, infatti, contribuirebbe a ridurre i danni dei radicali liberi. Un eccesso di questi sottoprodotti risulta infatti dannoso per le cellule, in quanto favorisce la produzione di forme tossiche di altre molecole, come i lipidi perossidati, inducendo lo stress ossidativo che, a sua volta, può portare alla degenerazione dei neuroni. In questo contesto è stato visto che la proteina tau gioca un ruolo fondamentale nella protezione dei neuroni dai lipidi danneggiati e/o perossidati favorendo la formazione di aggregati “tau-grassi” (droplet lipidici), che funzionano come un meccanismo di detossificazione.
Meccanismo d’azione
Quando i lipidi nelle membrane cellulari neuronali si danneggiano (perossidazione lipidica), la proteina Tau viene coinvolta nel loro “sequestro” associandosi ai lipidi danneggiati e formando delle goccioline specializzate (droplet). Questi droplets sono in grado, così, di isolare e di rimuovere i lipidi tossici riducendo il danno cellulare.
Tecniche di rilevazione delle proteine tau aggregate
La rilevazione dell’accumulo di tau è una sfida importante nella diagnosi e nel monitoraggio delle taupatie. Qui, di seguito, sono elencate le tecniche moderne più utilizzate:
- Imaging (tomografia a emissione di positroni);
- indagine condotta sul liquido cerebrospinale;
- microscopia elettronica;
- microscopia a forza atomica;
Tecniche avanzate di imaging, come la tomografia a emissione di positroni (PET), sono state sviluppate per visualizzare gli aggregati di tau in vivo. I traccianti PET specifici per la tau permettono di rilevare la distribuzione e la densità degli aggregati nel cervello. Questi traccianti si legano selettivamente agli aggregati di tau, fornendo immagini dettagliate che possono essere utilizzate per la diagnosi precoce e il monitoraggio della progressione della malattia.
Oltre all’imaging, l’analisi del liquido cerebrospinale (CSF) può rilevare livelli elevati di tau totale e tau fosforilata, che sono indicatori di neurodegenerazione.
Le tecniche di microscopia avanzata, come la microscopia elettronica e la microscopia a forza atomica, sono inoltre utilizzate per studiare la struttura degli aggregati di tau a livello molecolare. Queste tecniche forniscono informazioni dettagliate sulla morfologia e sulla composizione degli aggregati, contribuendo alla comprensione dei meccanismi di aggregazione.
Amiloidogenesi
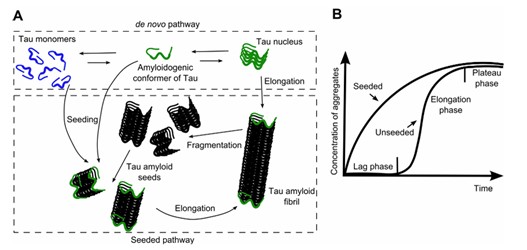
Gli aggregati amiloidi di Tau, come i PHF, si formano in un processo noto come amiloidogenesi o fibrillazione (Figura 2A). Nel corso di questo processo, molecole tau intrinsecamente disordinate si autoassemblano attraverso vari intermedi per formare fibrille amiloidi altamente ordinate e ricche in foglietti β. In particolare, nel processo di amiloidogenesi, alcune proteine tau, per un meccanismo ancora non chiaro, assumono una conformazione differente da quella fisiologica che viene definita “competente per l’amiloide”. In questa configurazione, la molecola tau si presenta ancora come singola proteina ma può iniziare ad aggregarsi. Più proteine Tau “competenti” si uniscono e formano un piccolo nucleo iniziale.
Cinetica di aggregazione
Formazione delle fibrille
Nella cinetica di polimerizzazione delle proteine tau, la formazione delle fibrille è considerata la fase più difficile e lenta (rate-limiting). Una volta formatosi il nucleo, infatti, altre singole proteine Tau saranno in grado di legarsi facilmente formando via via le fibrille amiloidi lunghe. Le estremità della fibrilla funzionano da molecole stampo così che nuove proteine tau possano essere indotte a cambiare forma e ad aggregarsi. Più estremità ci sono, maggiore sarà la velocità di crescita delle fibrille.
Rottura delle fibrille
Le fibrille neoformate possono andare incontro ad una rottura ed ogni singolo frammento diventa poi un “seme” da cui si genererà una nuova fibrilla. Il processo degenerativo diventa così sempre più veloce fino ad assestarsi in una fase di plateau (Figura 2B). Gli aggregati amiloidi possono, quindi, propagare le loro caratteristiche strutturali, portando alla conversione non fisiologica di altre molecole analoghe. Questo principio è alla base degli scenari di propagazione dei prioni nei mammiferi che causano encefalopatie spongiformi trasmissibili (TSE) note anche come malattie da prioni.
Diagnosi e trattamento
La diagnosi precoce e il monitoraggio dell’accumulo di proteine tau sono essenziali per intervenire tempestivamente e rallentare la progressione della malattia. Le terapie attuali per le taupatie sono principalmente sintomatiche. Tuttavia, la ricerca è attivamente impegnata nello sviluppo di terapie mirate che possano prevenire o ridurre l’aggregazione di tali molecole. Tra le strategie terapeutiche in fase di sviluppo vi sono gli inibitori delle chinasi, che mirano a ridurre la fosforilazione anomala delle proteine tau e gli anticorpi monoclonali che agiscono in modo mirato legandosi agli aggregati delle proteine tau e promuoverne la clearance.
Le terapie geniche rappresentano un’altra promettente area di ricerca: l’uso di tecnologie come CRISPR/Cas9 potrebbe infatti permettere di correggere mutazioni nel gene MAPT, prevenendo l’accumulo di tau patologiche. Questi approcci innovativi potrebbero rivoluzionare il trattamento delle taupatie, migliorando significativamente la qualità della vita dei pazienti.
{kind=link}
Fonti
- Tau Protein and Neurodegeneration: Michel Goedert | PDF | Neurodegeneration | Exon
- Proteina tau: nemica o alleata del cervello? – Brainer
- Amyloidogenesis of Tau protein – PMC
Fonti immagini
- Figura 1 – Tau Protein and Neurodegeneration: Michel Goedert | PDF | Neurodegeneration | Exon
- Figura 2 – Amyloidogenesis of Tau protein – PubMed