Lo spettro della sindrome di Zellweger (ZSD, Zellweger Spectrum Disease) è una malattia genetica rara causata da difetti nella biogenesi dei perossisomi.
Perossisomi disfunzionali in ZSD
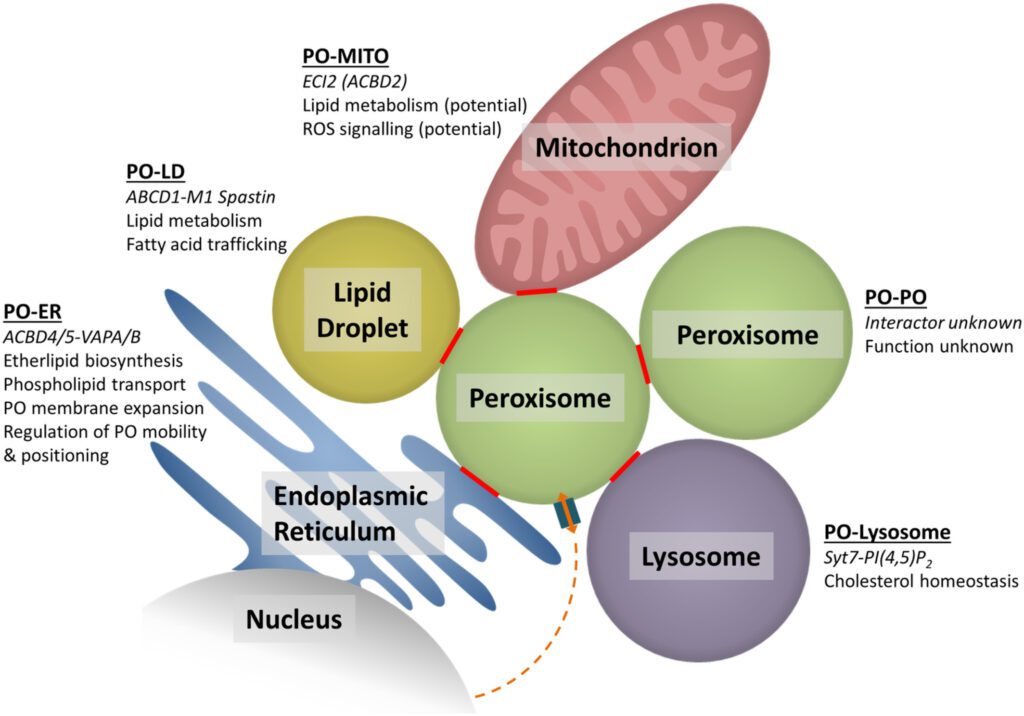
I perossisomi sono gli organelli cellulari predisposti a:
- la degradazione del perossido di idrogeno (H2O2);
- il metabolismo lipidico, in particolare con la β-ossidazione degli acidi grassi a catena lunga;
- la sintesi di acidi biliari.
Per svolgere correttamente queste funzioni i perossisomi cooperano e interagiscono con i mitocondri, il reticolo endoplasmatico e i lisosomi. I perossisomi sono centrali nel metabolismo cellulare di conseguenza se disfunzionali si accumulano metaboliti tossici.
Difetti nella biogenesi dei perossisomi
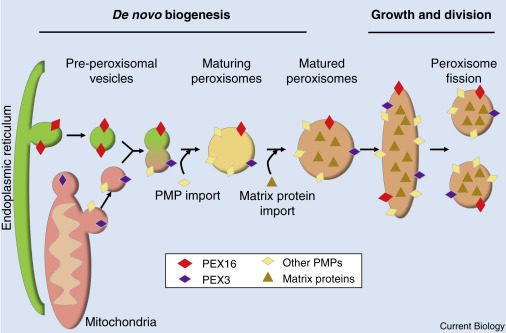
Esistono due processi che portano alla formazione dei perossisomi:
- la fissione, cioè divisione di perossisomi già esistenti;
- la biogenesi de novo dal reticolo endoplasmatico e mitocondri.
I disordini nella biogenesi dei perossisomi (PBDs, Peroxisome biogenesis disorders) sono malattie autosomiche recessive caratterizzate da difetti nella formazione della membrana, nell’importo delle proteine e/o nella proliferazione dei perossisomi. Pertanto questi organelli risultano disfunzionali e incapaci di compiere le proprie funzioni biochimiche. Lo spettro della sindrome di Zellweger (ZSD) fa parte dei PBDs, infatti è conosciuta anche come ZSD-PBD.
Nello specifico, ZSD è causata da mutazioni in uno dei 14 geni PEX che codificano per le perossine, le proteine necessarie per la formazione e il normale funzionamento dei perossisomi. Il gene più frequentemente interessato da mutazioni è PEX1 (70% dei casi), mentre mutazioni a PEX6, PEX10, PEX12 o PEX26 rappresentano un altro 26%. ZSD si trasmette con modalità autosomica recessiva, quindi occorre ereditare il gene mutato da genitori portatori sani per manifestare la malattia. Per individuare le mutazioni è utile l’analisi genetica tramite sequenziamento che permette di identificare individui sani portatori di mutazioni in eterozigosi e quindi la consulenza genetica per le famiglie.
Le malattie dello spettro di Zellweger
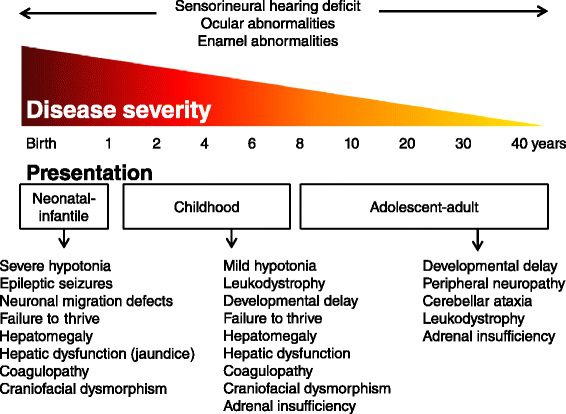
In ZSD l‘insorgenza e la gravità dei sintomi è molto variabile. I pazienti affetti presentano diversi livelli di gravità (severo, intermedio e lieve) dello stesso spettro clinico, per questo attualmente la malattia viene definita come uno spettro. Solitamente i pazienti con esordio precoce presentano il quadro più severo e una progressione più rapida, mentre nelle forme a esordio tardivo la presentazione clinica è più lieve e il decorso più lento.
Storicamente, sono state descritte tre diverse malattie dello spettro di Zellweger:
- sindrome di Zellweger (ZS, Zellweger syndrome), nota anche come sindrome cerebro-epato-renale, dal fenotipo severo;
- adreno-leucodistrofia neonatale (NALD, neonatal adreno-leukodystrophy) dal fenotipo intermedio;
- malattia di Refsum infantile (IRD, infantile Refsum disease) dal fenotipo più lieve.
Questa è una possibile classificazione, ma non è l’unica.
Gli individui affetti dallo spettro della sindrome di Zellweger possono presentare:
- dismorfismi craniofacciali (come viso appiattito, fronte alta e naso largo) e anomalie scheletriche;
- ritardo nella crescita;
- alterazioni e perdita della vista (deficit visivo, cataratta, glaucoma, retinite pigmentosa, atrofia del nervo ottico…);
- perdita dell’udito;
- sistema nervoso centrale (SNC) gravemente compromesso (difetti cerebrali e del midollo spinale, demielinizzazione);
- debolezza muscolare profonda;
- disfunzione epatica;
- disfunzione renale.
Diagnosi
Lo spettro della sindrome di Zellweger è generalmente sospettato clinicamente e poi confermato da analisi biochimiche e/o genetiche. ZSD può essere diagnosticato dimostrando alterazioni nelle funzioni biochimiche dei perossisomi, in particolare livelli plasmatici degli acidi grassi a catena molto lunga (VLCFA, very long chain fatty acids) alterati indicano metabolismo anomalo degli acidi grassi perossisomiali. In aggiunta, il sequenziamento dei 14 geni PEX è usato sempre più spesso come test di conferma ed è richiesto per malattie perossisomiali difficili da evidenziare con i tradizionali metodi biochimici.
Terapie
I meccanismi cellulari coinvolti in ZSD non sono noti, di conseguenza manca una terapia farmacologica efficace. Inoltre, l’ampia varietà nella sintomatologia, nella severità clinica e nella velocità di progressione aggiunge complessità al trattamento dei pazienti come singolo gruppo. Attualmente, la terapia è principalmente supportiva e personalizzata in base ai sintomi presenti. Indipendentemente dai trattamenti, la prognosi è sfavorevole e la maggior parte dei neonati affetti dalla sindrome di Zellweger muore nel primo anno di vita per la compromissione respiratoria inseguito a infezioni o epilessia intrattabile.
Fonti
- https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=IT&Expert=912
- https://www.ncbi.nlm.nih.gov/books/NBK1448/
- Wanders RJA, Baes M, Ribeiro D, Ferdinandusse S, Waterham HR. The physiological functions of human peroxisomes. Physiol Rev. 2023 Jan 1;103(1):957-1024. doi: 10.1152/physrev.00051.2021. Epub 2022 Aug 11. PMID: 35951481.
- Braverman NE, Raymond GV, Rizzo WB, Moser AB, Wilkinson ME, Stone EM, Steinberg SJ, Wangler MF, Rush ET, Hacia JG, Bose M. Peroxisome biogenesis disorders in the Zellweger spectrum: An overview of current diagnosis, clinical manifestations, and treatment guidelines. Mol Genet Metab. 2016 Mar;117(3):313-21. doi: 10.1016/j.ymgme.2015.12.009. Epub 2015 Dec 23. PMID: 26750748; PMCID: PMC5214431.
Crediti immagini
- Immagine in evidenza: https://www.biopills.net/wp-content/uploads/2016/11/perossisoma.jpg
- Figura 1: https://onlinelibrary.wiley.com/doi/10.1002/jimd.12083
- Figura 2: https://www.sciencedirect.com/science/article/pii/S0960982217302233
- Figura 3: https://ojrd.biomedcentral.com/articles/10.1186/s13023-015-0368-9