La fibrosi cistica, detta anche mucoviscidosi o malattia fibrocistica del pancreas è una patologia genetica che riguarda principalmente le ghiandole esocrine, quelle deputate alla produzione di muco e di sudore. Una volta istauratosi lo stato patologico, polmoni, pancreas, fegato, intestino, seni paranasali e apparato riproduttivo risentono maggiormente del malfunzionamento ghiandolare, determinando un quadro clinico davvero rilevante.
Epidemiologia
La fibrosi cistica è comunemente indicata come la patologia genetica grave più comune nella popolazione di etnia caucasica.
Si stima che in Italia 1 neonato su 2500 sia affetto da questa patologia, con circa 200 casi all’anno.
Negli Stati Uniti la condizione patologica riguarda circa 30000 individui, mentre nel Canada si stima che siano 3500 le persone affette. Con una visione un po’ più ampia, è possibile indicare 1 persona su 25 di discendenza europea e 1 su 30 di discendenza caucasica americana, è portatore di mutazioni responsabili di fibrosi cistica.
Controcorrente invece sembrano essere i dati provenienti dalle popolazioni di etnia differente. Infatti, solamente 1 bambino su 15000 afroamericani e 1 su 32000 negli asiatici-americani mostra la patologia.
L’incidenza più elevata al mondo è però presente in Irlanda con una stima di 1 caso su 1353 individui.
Non è una malattia di genere, in quanto si riscontra ugualmente in maschi e femmine, anche se la mortalità sembra mostrare un numero maggiore nel genere femminile. La spiegazione non è ancora chiara, ma alcuni studi mostrano una correlazione dell’ormone femminile estrogeno con la gravità della patologia.
La fibrosi cistica è una malattia autosomica recessiva, quindi mostreranno la piena patologia solamente individui omozigoti per il gene responsabile. Infatti, gli eterozigoti avranno un fenotipo funzionale, anche se con qualche particolarità.
Patogenesi e fattori di rischio
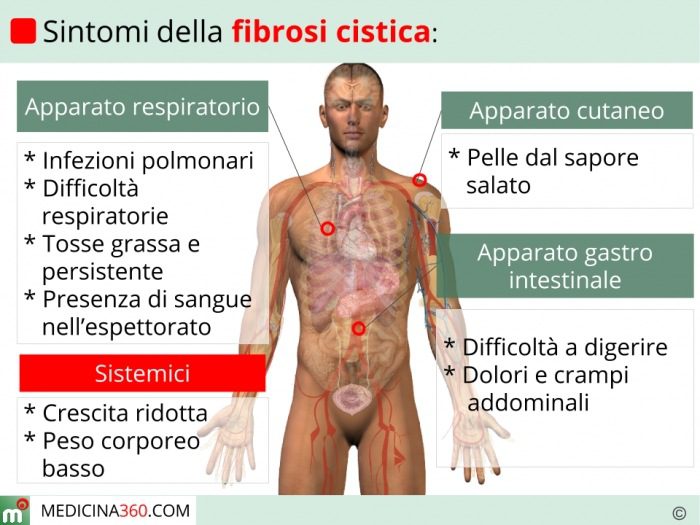
Le manifestazioni cliniche sono varie e differiscono anche per l’organo o l’apparato che interessano. Innanzitutto, i segni evidenti della presenza patologica sono la caratteristica pelle salata, la crescita ridotta o assente, l’accumulo del caratteristico muco denso, viscoso e appiccicoso che porta frequentemente ad infezioni polmonari, tosse e dispnea. Inoltre, la fibrosi cistica può determinare, in rari casi, problemi relativi alla coagulazione del sangue. Nei bambini si riscontrano disturbi da malassorbimento di vitamina K, strettamente collegati alla coagulazione sanguigna.
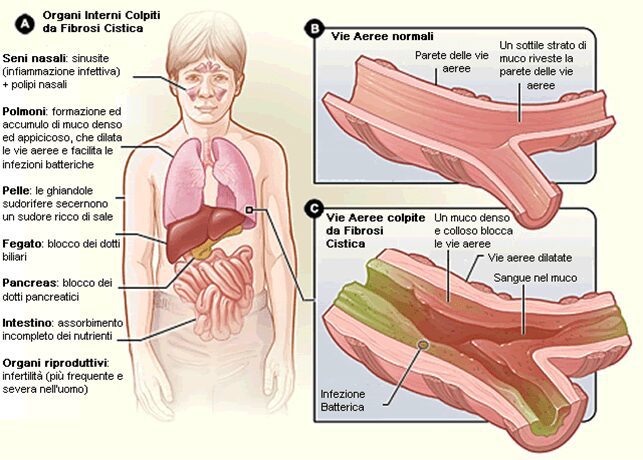
Il malfunzionamento ghiandolare, quindi, determina una produzione di muco molto elevata, con ripercussioni a vari livelli. A livello polmonare l’accumulo di muco determina ostruzione delle vie aeree, riduzione della motilità ciliare e infiammazione. Successive a questa fase sono varie infezioni opportunistiche, che determinano anche lesioni e cambiamenti strutturali polmonari.
A livello gastrointestinale, nei neonati, si riscontra l’impossibilità di espellere feci (meconio), con possibile prolasso rettale.
La presenza del muco determina il blocco della secrezione di enzimi digestivi ed i loro movimenti nel duodeno, portando a pancreatite e ad atrofia dei dotti pancreatici. Determinando, inoltre, malassorbimento nutritivo data l’assenza degli enzimi. Le stesse peculiarità, con la produzione di bile, si riscontrano nel fegato. La bile può bloccare i dotti biliari con danni epatici e cirrosi.
Nel pancreas, inoltre, può venir meno la produzione insulinica ed anche la perdita di funzionalità delle cellule di Langerhance, portando a diabete.
Infine, può presentarsi infertilità sia maschile che femminile.
La patologia sarà presente solamente in omozigosi, quindi quando si incontrano due genitori portatori del gene avranno il 25% di probabilità di avere un figlio malato.
Descrizione anomalia genetica
Cystic Fibrosis Transmembrane Regulator (CFTR) è il gene responsabile della comparsa della fibrosi cistica. Si trova nel locus q31.2 del cromosoma 7, è strutturato da 230.000 paia di basi e codifica una proteina composta da 1480 aminoacidi. Le mutazioni presenti sul gene CFTR determinano la presenza/assenza e l’entità della patologia. La mutazione più frequentemente presente è quella che riguarda la perdita di 3 paia di basi nell’esone 10, comunemente denominata “Delta-F508”. Questa delezione si traduce nella perdita della fenilalanina nella posizione 508. Il gene CFTR codifica per una proteina facente parte della famiglia delle ATPasi di traffico, ovvero un trasportatore, deputata al trasporto dello ione cloro. Quindi, la proteina codificata non sarà in grado di trasportare attivamente cloro e indirettamente sodio e acqua. La mancata eliminazione di acqua secondo un gradiente osmotico determina la creazione di muco eccessivamente denso e viscoso.

La Delta-F508 non rappresenta però l’unica mutazione riscontrata. Attualmente sono riscontrate altre 1500 ulteriori mutazioni in grado di determinare la patologia.
Tuttavia, è presente una grande divergenza tra genotipo e fenotipo. Ovvero le manifestazioni e la gravità della malattia dipendono molto dal tipo di mutazione presente. Non tutte le mutazioni e le forme della malattia sono gravi, ma ne esistono molte forme lievi che non determinano il quadro clinico estremo.
E’ una malattia autosomica recessiva, per cui solo l’omozigosi determinerà un quadro patologico, a diversi livelli. L’omozigosi Delta F508/ Delta F508 è considerata la forma “classica” della patologia, e sembra mostrare ancora la pericolosità maggiore.
Caratteristiche cliniche e complicanze
Bronchiectasia e polipi nasali sono due tra le principali complicanze dettate dalla fibrosi cistica a livello respiratorio. Inoltre, Staphylococcus aureus, Haemophilus influenzae e Pseudomonas aeruginosa sono i microorganismi più frequentemente riscontrati tra le infezioni opportunistiche successive ad ostruzione delle vie aeree. Il quadro clinico viene ulteriormente aggravato da tosse con sangue (emottisi), insufficienza cardiaca e respiratoria.
Nel sistema digerente invece si presentano carenze nutrizionali significative, dati i blocchi dei dotti pancreatici che impediscono l’assorbimento dei macronutrienti.
L’assenza di insulina porta molto frequentemente a diabete anche prima dei 30 anni.
Nei bambini, inoltre, esiste il rischio di intussuscezione che va a determinare occlusione intestinale.
Infine la sterilità riproduttiva nei maschi è data dall’ostruzione dei dotti deferenti. Nelle donne la sterilità è meno frequente, infatti alcune mantengono la possibilità di portare avanti una gravidanza. La gravidanza però potrebbe aggravare ulteriormente i sintomi della fibrosi cistica.
Diagnosi
La diagnosi prenatale ha assunto un ruolo sempre più decisivo per la lotta alla fibrosi cistica. Con le analisi genetiche è possibile innanzitutto testare entrambi i genitori prima di una gravidanza, in modo determinare la presenza di mutazioni sul gene CFTR. Si raccomanda che questo test venga eseguito su almeno un genitore, data la condizione di omozigosi per la manifestazione patologica. In più, il test può essere eseguito in gravidanza, sulla placenta o sul liquido amniotico, tramite il prelievo dei villi coriali oppure l’amniocentesi.
Va ricordato il test del sudore che può essere eseguito come test neonatale, o come screening sulla popolazione. Il sudore raccolto viene analizzato per valori maggiori di sodio e cloro. Infine, si può analizzare la saliva per la minor presenza di tiocianato e isotiocianato.
Trattamento e prevenzione
Rispetto al passato sono stati fatti enormi passi avanti per il trattamento della fibrosi cistica. Basti pensare che fino a più di mezzo secolo fa, i bambini affetti, a stento superavano l’anno di età mentre oggi possono raggiungere l’età adulta. La gestione patologica riguarda ogni suo aspetto, con terapie polmonari, gastrointestinali, pancreatiche, riproduttive ed anche di supporto psicologico. Non si tratta, però di una cura a tutti gli effetti contro la patologia, ad oggi ancora non disponibile.
I polmoni e le vie aeree restano il punto principale dei trattamenti. Gli antibiotici svolgono un ruolo fondamentale contro le infezioni secondarie e croniche, mentre dispositivi meccanici di inalazione sono necessari per eliminare il muco in eccesso.
In casi estremi, si rende necessario il trapianto degli organi colpiti, partendo dal trapianto polmonare fino a quello del fegato.
Per il diabete causato dal deficit insulinico si ricorre a iniezioni o all’uso di una pompa di insulina.
A seconda delle manifestazioni cliniche, verrà stabilito un trattamento ideale, anche se non specifico.
Per questa ragione si sta percorrendo la strada della terapia genica per ristabilire la funzionalità di CFTR. Tramite due metodi: l’utilizzo di un vettore virale (adenovirus) oppure grazie all’uso di liposomi. Da qualche anno però la Crispr/Cas9 ha aperto nuove frontiere di ingegneria genica, per cui la ricerca può davvero considerare l’idea di attuare un cambiamento totale del gene, ristabilendo la condizione non patologica.
Fonti
- www.fibrosicistica.it/fibrosi-cistica/cose-la-fibrosi-cistica/
- www.osservatoriomalattierare.it/malattie-rare/fibrosi-cistica
- www.fibrosicisticaricerca.it/cose-la-fibrosi-cistica/
- www.humanitas.it/malattie/fibrosi-cistica/
- www.my-personaltrainer.it/salute/fibrosi-cistica.html
- www.telethon.it/cosa-facciamo/ricerca/malattie-studiate/fibrosi-cistica/
- https://www.medicina360.com/fibrosi-cistica.html
- http://malattierare.sanita.basilicata.it/FibrosiCistica.asp
- https://www.cdi.it/approfondimento/la-fibrosi-cistica/
- https://it.blastingnews.com/salute/2017/04/la-timosina-a1-e-i-suoi-effetti-positivi-sulla-fibrosi-cistica-001643077.html
I commenti sono chiusi.