Caratteristiche
L’Herpes virus umano 8 (HHV-8) o virus associato al sarcoma di Kaposi (KSHV) è un virus a DNA a doppio filamento di circa 165 kilobasi, con capside a simmetria icosaedrica, rivestito (Fig. 1), appartenente alla famiglia Herpesviridae e alla sottofamiglia dei gamma herpes virus. In quest’ultima sono raggruppati i virus erpetici che hanno un tropismo limitato, si replicano lentamente e sono in grado di immortalizzare le cellule trasformandole in cellule tumorali (qui rientra anche il virus di Epstein-Barr, agente eziologico della mononucleosi e di altre patologie).

La scoperta di questo virus risale al 1994 grazie al lavoro di due ricercatori, Yuan Chang e Patrick S. Moore, i quali lo identificarono in biopsie di un tumore chiamato sarcoma di Kaposi; questo era già noto dalla seconda meta del XIX secolo e il numero delle persone colpite incrementò fortemente in seguito alla diffusione dell’AIDS. Questi due scienziati, contrariamente alla comunità scientifica, erano convinti che la causa del sarcoma di Kaposi non era l’HIV, dal momento che non tutti gli individui HIV positivi sviluppavano questa neoplasia. Chang e Moore identificarono il virus grazie all’impiego di una tecnica chiamata analisi della differenza rappresentazionale (RDA), che consente di mettere a confronto i genomi di cellule diverse combinando un’ibridazione sottrattiva a un’amplificazione tramite PCR; essi individuarono una sequenza di circa 180 bp (presente nel 90% dei campioni di sarcoma di Kaposi provenienti da individui con AIDS), sulla quale effettuarono il sequenziamento e l’analisi bioinformatica dimostrandone l’origine non umana e l’omologia con i geni della proteina del tegumento e del capside minore dei gamma herpes virus (Epstein–Barr virus e herpes virus saimiri). Tale rivelazione portò alla definizione di un nuovo herpes virus umano; oltre a ciò, Chang e Moore notarono che questa sequenza era presente in tutte le forme di sarcoma di Kaposi.
È importante evidenziare che il sequenziamento del KSHV ha permesso di svelare una caratteristica straordinaria riguardante il suo genoma, ovvero la somiglianza dei suoi prodotti genici con molte proteine umane coinvolte nella sopravvivenza cellulare, nella proliferazione e nell’angiogenesi (Fig. 2). Inoltre, il genoma dell’HHV-8 codifica per svariate proteine che agiscono da inattivatori dei geni oncosoppressori; un esempio è LANA, di cui parleremo più avanti.
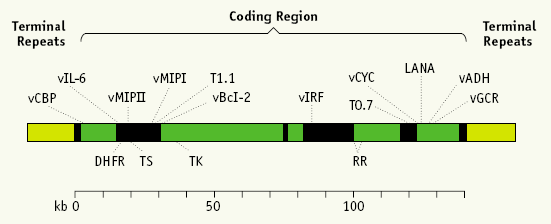
Rispetto agli altri herpes virus, l’HHV-8 non è ubiquitario, bensì presenta una sieroprevalenza variabile nelle diverse aree del mondo: in Nord Europa e Stati Uniti d’America è 1-5%, nella popolazione omosessuale del Nord Europa e degli USA 40%, nell’area mediterranea 10-30%, e nell’Africa sub-sahariana più del 40% (Fig. 3).

Filogenesi
Dominio Duplodnaviria
Regno Heunggongvirae
Phylum Peploviricota
Classe Herviviricetes
Ordine Herpesvirales
Famiglia Herpesviridae
Sottofamiglia Gammaherpesvirinae
Genere Rhadinovirus
Specie Human gammaherpesvirus 8
Replicazione
L’HHV-8 infetta diversi tipi cellulari, compresi endoteliociti, linfociti B, cellule epiteliali, cellule dendritiche, monociti e fibroblasti. Si pensa che, per entrare nelle cellule endoteliali, l’HHV-8 si leghi a molteplici recettori espressi sulla membrana, come le integrine, il trasportatore cistina-glutammato (xCT), il grande trasportatore di amminoacidi neutri (CD98) l’eparan solfato e il recettore a tirosina chinasi EPHA2; questo legame attiva una cascata di trasduzione di segnali che comporta cambiamenti cellulari che consentono al virus di penetrare nella cellula e muoversi nel citoplasma. L’ingresso del virus risulta nella rimozione del pericapside virale (uncoating), nel rilascio del capside nel citoplasma, seguito dal suo trasporto verso la membrana nucleare e dalla liberazione del genoma virale nel nucleo, all’interno del quale diventa circolare e rimane in forma episomale (Fig. 4). A questo punto il virus entra nello stato di latenza oppure inizia il ciclo litico.
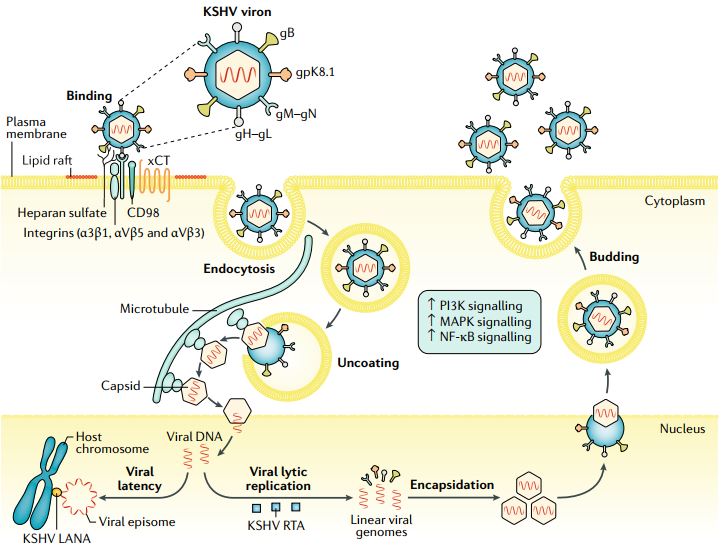
Come per altri herpes virus, l’infezione da parte del KSHV può durare tutta la vita, in quanto il virus stabilisce una latenza nei linfociti B e nelle cellule endoteliali; durante questa fase del ciclo, il virus esprime i seguenti geni della latenza:
- ORF7, che codifica per vFLIP (viral Fas-associated protein with death domain-like interleukin-1β-converting enzyme/caspase-8-inhibitory protein), che inibisce l’apoptosi e attiva il pathway di NF-kB, responsabile della sopravvivenza cellulare e della secrezione di citochine proinfiammatorie (IL-6 e IL-8);
- ORF72, che codifica per la Ciclina virale (vCyc), un omologo della Ciclina D2 cellulare, che induce la proliferazione;
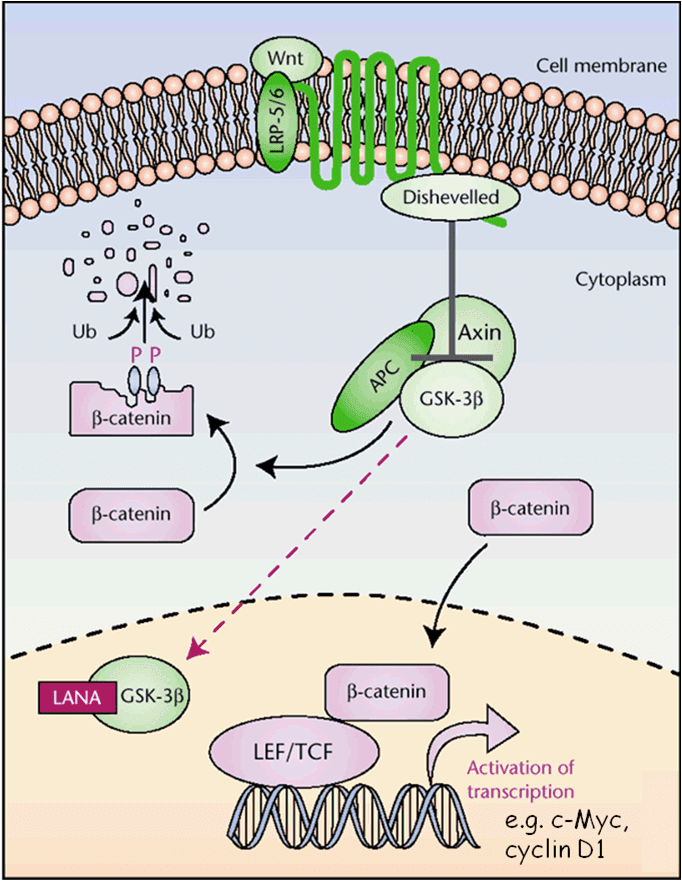
- ORF73, che codifica per LANA, la principale proteina di latenza. Questa lega e inibisce i geni oncosoppressori p53 e RB, e attiva il pathway di Wnt-β-catenina (Fig. 5), stimolando le cellule ad entrare nella fase S della mitosi. In aggiunta a ciò, àncora gli episomi virali ai cromosomi della cellula ospite, consentendone la segregazione nelle cellule figlie durante la mitosi;
- ORFK12, che codifica per le kaposine, proteine di segnale che aumentano il rilascio di citochine proinfiammatorie;
- Diversi microRNA, tra cui miR-K12-3, che promuove la migrazione e l’invasione delle cellule endoteliali attivando la proteina chinasi B (AKT), e miR-K9, che ha come bersaglio il trascritto della maggiore proteina del ciclo litico (ORF50), al fine di prevenire la riattivazione dalla latenza.
Per quanto riguarda il ciclo litico, gli stimoli fisiologici che permettono la riattivazione spontanea dell’HHV-8 dalla latenza non sono ancora ben definiti. Tuttavia, è chiaro che il virus va incontro alla riattivazione del ciclo litico in maniera sporadica per tutta la durata della vita dell’ospite. La fase litica, durante la quale i geni virali sono espressi in un ordine temporale, permette la replicazione del genoma virale e la produzione della progenie virale infettante. I geni immediatamente precoci vengono espressi per primi, e in questi rientra la proteina litica chiave, RTA, che è un fattore di trascrizione che attiva molti geni virali e i promotori cellulari, e assicura l’espressione dei geni virali richiesta per la replicazione del virus. I geni precoci ritardati e i geni tardivi sono espressi dopo i geni precoci. La maggioranza dei geni precoci ritardati codifica per proteine che controllano la replicazione del DNA virale, che avviene attraverso un processo di replicazione unidirezionale e porta alla sintesi di genomi lineari che vengono racchiusi nei capsidi. La fase litica tardiva è caratterizzata dall’espressione di tutte le proteine virali strutturali e dalla produzione dei nuovi virus infettanti.
Va evidenziato come sia le proteine codificate dai geni di latenza che le proteine codificate dai geni del ciclo litico possono contribuire alla tumorigenesi. Ciononostante, diversamente dai geni latenti, che sono espressi in tutte le cellule tumorali, i geni del ciclo litico sono espressi solo in una piccola quantità di cellule cancerose. Alcune proteine del ciclo litico possono promuovere la crescita tumorale mediante meccanismi paracrini, cioè rilasciando dei segnali chimici che raggiungono le cellule bersaglio (è diverso dal segnale endocrino, in cui i messaggeri chimici entrano nel circolo ematico). L’esempio primario è vGPCR (viral G-protein-coupled receptor), una proteina transmembrana omologa al recettore dell’interleuchina 8, in grado di indurre costitutivamente la segnalazione cellulare favorendo l’espressione di fattori proinfiammatori e angiogenici (come il fattore di crescita dell’endotelio vascolare, VEGF, e il fattore di crescita derivato dalle piastrine, PDGF), con conseguente aumento della sopravvivenza cellulare, dell’angiogenesi e della proliferazione (Fig. 6). Secondo un lavoro del 2003, pubblicato su Cancer Cell, la proteina vGPCR è espressa solo nel 10% delle cellule tumorali, ma riesce a promuovere la cancerogenesi mediante il rilascio di VEGF, che agisce sulle cellule esprimenti i geni della latenza consentendo la proliferazione.

Date le sue proprietà, vGPCR raffigura un bersaglio terapeutico ideale per il trattamento del sarcoma di Kaposi. Questo è stato descritto in uno studio pubblicato su Cancer Research nel 2006, in cui i ricercatori produssero delle cellule endoteliali esprimenti vGPCR sensibili all’antivirale Ganciclovir in seguito alla co-espressione della timidina chinasi del virus herpes simplex 1, che attiva il farmaco. Nei modelli murini si è visto che per indurre la regressione del tumore era sufficiente colpire le poche cellule positive a vGPCR. Le cellule restanti, esprimenti i geni latenti, non erano capaci di garantire la crescita del tumore in assenza dei segnali paracrini provenienti dalle cellule vGPCR+.
Patogenesi
Il KSHV è responsabile di varie patologie, di cui la più comune è il sarcoma di Kaposi (KS) che fu descritto per la prima volta nel 1872 dal dermatologo ungherese Moritz Kaposi come una neoplasia angiomatosa. In quel periodo Kaposi lavorava al dipartimento di dermatologia dell’università di Vienna, e inizialmente chiamò questo tumore “sarcoma pigmentato multiplo idiopatico della pelle”.
Il KS è un tumore mesenchimale multifocale che si genera dalle cellule della linea dei vasi sanguigni e di quelli linfatici, quindi coinvolge le cellule endoteliali. L’istopatologia e l’andamento clinico sono altamente eterogenei, la componente infiammatoria è un cofattore essenziale per il suo sviluppo, in quanto il virus è necessario ma non sufficiente.
Il KS consiste in un’angiogenesi intensa ed aberrante, con cellule proliferanti dette “spindle cell”, ossia cellule fusiformi che creano dei vasi sanguigni irregolari e discontinui che permettono la fuoriuscita delle cellule del sangue, specialmente gli eritrociti (Fig. 7A e 7B). Tale evento è alla base del colore rosso-violaceo delle lesioni cutanee tipiche in cui c’è un’abbondanza di globuli rossi che muoiono e rilasciano emoglobina, che si ossida. All’interno di queste lesioni si trovano le spindle cell (Fig. 7C), molte delle quali contengono il virus latente; tuttavia, data la natura infiltrativa di tali lesioni, le cellule non infette, che non albergano il virus, sono mischiate alle cellule tumorali. Infatti nelle lesioni tipiche del KS risiedono molteplici tipi cellulari, tra cui cellule endoteliali, spindle cell con il virus latente, spindle cell con il virus in fase litica, spindle cell con disregolazione del programma trascrizionale, cellule immunitarie (soprattutto monociti e macrofagi), globuli rossi e cellule progenitrici endoteliali (EPC). In più ci sono le citochine, importanti per la crescita del tumore.
Le lesioni cutanee del KS variano nel corso della malattia seguendo gli stadi di evoluzione: all’inizio appaiono come delle macchie (o macule) rossastre, piatte o rialzate, e indolori, dopo si trasformano in placche edematose violacee che si induriscono, e infine diventano noduli, che raffigurano la fase più aggressiva, in cui c’è un’espansione clonale delle spindle cell (Fig. 8). Quando si arriva a questo stadio, il 100% delle spindle cell sono positive all’HHV-8, e solo il 10% ospita il virus in fase litica. Bisogna ricordare che sia il ciclo latente sia il ciclo litico contribuiscono alla patogenesi del KS.

Esistono quattro tipologie di KS:
- Classico, che colpisce principalmente gli uomini anziani, dal momento che il recettore degli androgeni permette l’attivazione di uno dei recettori cellulari del virus facilitandone l’endocitosi. È diffuso soprattutto in Italia, Grecia, Turchia e Israele, si manifesta con placche rosso-violacee o piccoli noduli alle estremità (Fig. 9B, 9C, 9G), metastatizza raramente e non porta alla morte;
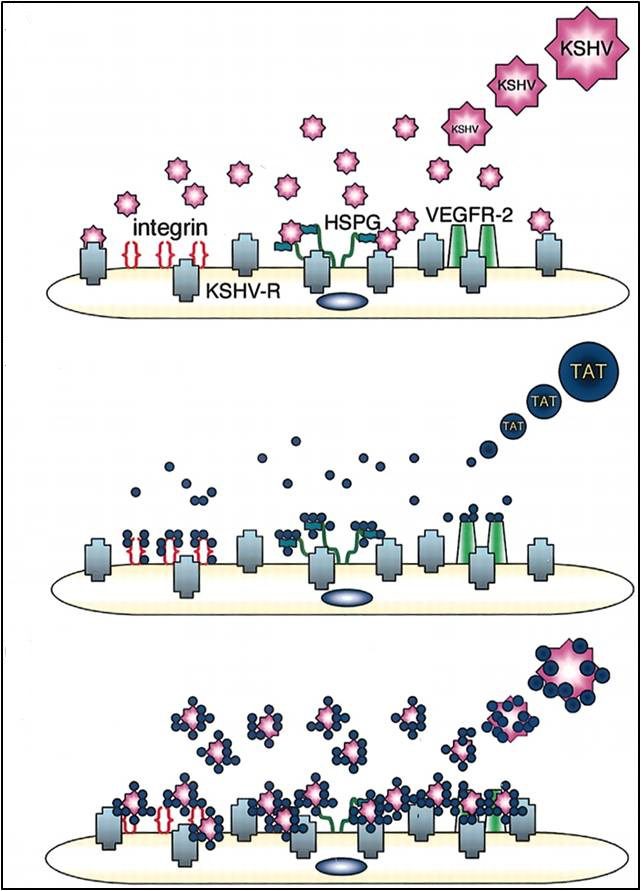
- Associato all’AIDS, che colpisce gli individui con il sistema immunitario compromesso dall’HIV, ed è quello più fatale. È caratterizzato da lesioni che progrediscono velocemente verso i noduli, che compaiono sul viso, sul busto, sul palato e sulle gengive (Fig. 9A, 9D, 9E, 9F). Inoltre, si diffonde agli organi interni, specie quelli ricchi di vasi linfatici, come i polmoni. È stato proprio questo tipo di tumore a provocare la morte del musicista e cantautore Freddie Mercury il 24 novembre 1991. La correlazione tra HIV e la progressione del KS sta non solo nell’immunodepressione, ma anche nell’azione della proteina Tat dell’HIV, che si lega al recettore del KSHV, funge da collante e concentra il virus (Fig. 10);
- Endemico, tipico dell’Africa sub-sahariana, che è clinicamente simile a quello classico, ma colpisce maschi più giovani (dai 35 anni). Esiste un sottotipo più letale e fulminante che coinvolge i bambini molto piccoli, non presenta lesioni cutanee ma si propaga rapidamente ai linfonodi e agli organi interni;
- Iatrogeno, associato al trapianto di organi, la cui incidenza varia a seconda dell’area geografica (0,4% negli USA e in Europa, 87,5% in Arabia Saudita, 80% in Turchia). La malattia può essere dovuta alla riattivazione del KSHV in riceventi sieropositivi (che si sottopongono a terapie immunosoppressive prima del trapianto), oppure alla trasmissione del virus attraverso il trapianto in riceventi sieronegativi. È contraddistinto da lesioni cutanee e alle mucose, e di rado si estende agli organi viscerali.

Altre due patologie, meno abituali, provocate dal KSHV sono i linfomi primitivi effusivi (PEL) e i disordini multicentrici di Castleman (MCD). I PEL sono dei linfomi monoclonali a cellule B, non Hodgkin, rari e rapidamente mortali, che si estendono nelle cavità sierose, ovvero il peritoneo, le pleure e il pericardio; si chiamano così perché non è presente una massa tumorale solida individuabile. Nei PEL è frequente una co-infezione con il virus di Epstein-Barr (nell’80% dei casi). Nonostante derivino dai linfociti B, questi tumori spesso non esprimono immunoglobuline o altri antigeni delle cellule B, bensì sono positivi ai marcatori delle plasmacellule e presentano una morfologia centroblastica/immunoblastica (Fig. 11)
Per quanto concerne gli MCD, si tratta di malattie linfoproliferative a cellule B, aventi una patogenesi eterogenea con solo il 50% di casi dovuti all’infezione da HHV-8 in pazienti sani e negativi all’HIV. La causa degli individui negativi al KSHV e colpiti dagli MCD rimane sconosciuta, ma quasi certamente è connessa alla disregolazione della secrezione dell’interleuchina 6. Contrariamente ai PEL, gli MCD sono caratterizzati da espansione policlonale in cui gran parte della componente cellulare è formata da linfociti non infettati reclutati nel sito dell’infezione dalle citochine prodotte da una ristretta quantità di cellule positive al KSHV. C’è un alto tasso di linfomi non Hodgkin secondari tra i pazienti con MCD, e ciò suggerisce che questo disordine può predisporre o evolvere verso un disordine neoplastico. A seconda dell’aspetto microscopico, gli MCD sono descritti come ialini-vascolari, misti e plasmablastici (Fig. 12).


Metodi di identificazione
Quando c’è il sospetto clinico di KS, si preleva una biopsia tissutale al fine di confermare la diagnosi a livello istologico. Anche se tale procedura è facile in ambienti ricchi di risorse, può essere ardua in ambiente con risorse limitate, per esempio in Africa, dove il KS è più comune, e l’analisi clinica macroscopica è spesso l’unico mezzo disponibile per la diagnosi di questo tumore. Infatti, in uno studio proveniente dall’Africa orientale, la sola diagnosi visiva ha solamente l’80% di valore predittivo positivo per il KS; alcuni pazienti sono risultati falsi positivi, e perciò è stata loro indicata una chemioterapia non necessaria. Per rimediare a tali inconvenienti, vari sforzi hanno avuto come scopo quello di aumentare la diagnosi istologica del KS, compreso lo spostamento dell’esecuzione delle biopsie anche ai non medici usando la teledermatologia e la telepatologia.
La diagnosi patologica di KS spesso può essere eseguita mediante la convenzionale colorazione ematossilina-eosina (H&E) da sola, al fine di valutare diverse caratteristiche basilari del tumore che sono presenti, a vari livelli, in tutti i casi della malattia. Queste proprietà comprendono la proliferazione vascolare nel derma (con la genesi di spazi simili a tagli che non sono ricoperti dall’endotelio), un incremento del numero dei vasi senza una linea cellulare endoteliale, la presenza di extravasazione del sangue con formazione di globuli ialini, accumulo di emosiderina e infiltrato infiammatorio (Fig. 13). La proliferazione delle spindle cell è un altro aspetto tipico del KS; tali cellule, che sono caratterizzate da citoplasma e nucleo allungati, e qualche volta contengono emosiderina e inclusioni ialine, esprimono marcatori endoteliali (CD34, VEGFR) e sono considerate le cellule tumorali del KS. Nonostante le spindle cell siano di solito raggruppate in fogli o in fasci (Fig. 13B), possono essere difficili da distinguere nelle lesioni precoci (Fig. 13C).
Le macule di KS possono essere le più difficili da distinguere istologicamente da altre condizioni, poiché possono mimare altri disordini infiammatori della cute, come anomalie vascolari minori (Fig. 13C e 13D). Le macule di KS sono contraddistinte da un infiltrato perivascolare sparso composto da linfociti e plasmacellule, dalla presenza di eritrociti fuoriusciti dai vasi, macrofagi contenenti ferro (siderofagi), fili ristretti di cellule tra fasci di collagene, e qualche volta fasci di spindle cell (Figura 7A).
Le placche di KS presentano un infiltrato diffuso di vasi in ogni parte del derma, con fasci di spindle cell che rimpiazzano il collagene. Gli spazi vascolari di solito hanno contorni seghettati e fasci di collagene separati. È comune l’extravasazione dei globuli rossi con siderofagi; l’infiltrato infiammatorio comprende numerosi macrofagi, linfociti e plasmacellule (Fig. 7B).
Le lesioni nodulari di KS sono quelle istologicamente più definite dal momento che sono caratterizzate da noduli ben definiti formati da fogli di spindle cell che sostituiscono il collagene dermico (Fig. 13E). Un pattern di spazi vascolari, somigliante a un nido d’ape e pieno di eritrociti, è frequentemente associato con le spindle cell che si intrecciano. Sono ricorrenti gli spazi perivascolari dove le emazie appaiono a diretto contatto con le spindle cell. Anche in queste lesioni si nota l’extravasazione degli eritrociti con siderofagi e globuli ialini; questi ultimi sono delle sfere eosinofile di 1-7 μm di diametro. I casi avanzati di KS, come la variante anaplastica, possono rivelare fogli di spindle cell atipiche che mimano altri sarcomi; in questi casi l’analisi immunoistochimica (IHC) aiuta la diagnosi.
Le lesioni di KS hanno una composizione cellulare eterogenea; l’IHC sulle spindle cell, utilizzando anticorpi specifici per i marcatori endoteliali, ne svelò la natura vascolare e la conseguente individuazione di marcatori endoteliali linfatici, come podoplanina, LYVE1 e VEGFR3 (recettore 3 di VEGF), dimostrando come il KS abbia un’origine cellulare linfatica endoteliale. Tuttavia le lesione di KS esprimono anche marcatori di origine mesenchimale, come la vimentina, e per questo è stata recentemente proposta l’origine mesenchimale del KS. La maggioranza delle evidenze immunoistochimiche, l’espressione genica e i dati sperimentali attualmente suggeriscono che le spindle cell siano cellule linfatiche endoteliali, endoteliali vascolari e/o mesenchimali che vanno incontro alla riprogrammazione che segue l’infezione da KSHV, al fine di produrre cellule con un immunofenotipo combinato aberrante. In aggiunta ai linfociti e alle plasmacellule, anche gli istiociti sono abbondanti nelle lesioni di KS, e possono essere identificati mediante IHC con anticorpi che riconoscono i loro marcatori: CD1a, CD207, S100.
Le colorazioni immunoistochimiche per gli antigeni del KSHV sono assai utili nella diagnosi di KS; in particolare, gli anticorpi che riconoscono LANA possono essere impiegati nell’istopatologia di routine per confermare la diagnosi di KS nei casi complicati. L’IHC specifica per LANA è alquanto vantaggiosa per diagnosticare i casi di KS con lesioni maculari precoci o lesioni che ricordano altri sarcomi. Comunque, nonostante si pensi che LANA sia espressa in ogni cellula infettata, la proporzione di cellule alberganti il virus è variabile, e oscilla da <10% a >90% della popolazione cellulare totale nelle aree delle lesioni (Fig. 13D, 13F).
Inoltre, l’IHC per LANA dovrebbe essere considerata positiva solo quando è visibile un pattern nucleare puntinato, che impedisce alla colorazione di LANA di essere confusa con emosiderina citoplasmatica o melanina quando si usa un cromogeno marrone. Malgrado la positività di LANA confermi la diagnosi di KS, una colorazione negativa può non escludere il KS, poiché falsi negativi possono essere dovuti a scarsa conservazione del tessuto o ad altri artefatti tecnici. Quindi, una colorazione positiva per LANA è richiesta allo scopo di stabilire la diagnosi di KS e dipende da specifiche circostanze.

L’identificazione del DNA di KSHV mediante PCR è altamente sensibile per il KS, e l’assenza del DNA virale in un campione ben preparato essenzialmente esclude la diagnosi. Tuttavia, eccetto per i laboratori di ricerca, la PCR specifica per l’HHV-8 è attualmente disponibile solo in pochi laboratori di patologia molecolare clinica specializzati. La specificità dell’individuazione del DNA di KSHV è meno chiara soprattutto nelle regioni dove il virus è endemico, in cui fino al 60% delle persone nella comunità risultano positive agli anticorpi anti-KSHV. A tal proposito i dati sono parecchio limitati (il 14% dei campioni in archivio da individui senza il KS in Uganda erano positivi per il DNA del KSHV), però suggeriscono che una soglia quantitativa del DNA di HHV-8 può distinguere le persone positive al DNA virale con KS da quelle positive che non hanno il KS. Se è così, la diagnosi molecolare obiettiva automatica del KS potrebbe essere eseguita accanto alla cura clinica e, in larga parte, rimuovere la soggettività e le spese della diagnosi istopatologica. Un nuovo dispositivo da usare per la diagnosi di KS che presenta multiple sorgenti di energia per l’uso quando l’elettricità è limitata, è in via di sviluppo. L’ultimo stadio di questo dispositivo, chiamato TINY (Tiny Isothemal Nucleic acid quantifications sYstem), si affida all’amplificazione isotermica e può essere scaldato tramite elettricità, luce del sole o fiamme. Il test su tale apparecchio in Uganda mostrò che il DNA di KSHV potrebbe essere determinato in campioni di biopsie di KS in meno di tre ore dopo aver applicato l’anestesia al paziente. La vasta implementazione di questo congegno potrebbe consentire una diagnosi definita del paziente mentre si trova ancora in ambulatorio; invece, quando si effettua la diagnosi patologica, costui deve aspettare diverse settimane. Un dispositivo come TINY può portare alla diagnosi più precoce e prevenire la perdita di follow-up.
Terapia
Nei pazienti affetti da forme di KS in cui l’immunosoppressione è potenzialmente reversibile, l’approccio di prima linea è quello di rinforzare il sistema immunitario, per esempio mediante il trattamento dell’HIV con il protocollo terapeutico HAART (Highly Active Antiretroviral Therapy) negli individui con KS associato all’AIDS. Questo trattamento permette la regressione del tumore allo stadio T0. In maniera simile, i pazienti con KS iatrogeno possono essere trattati riducendo il livello di immunodepressione o cambiando i farmaci immunosoppressori utilizzati (per esempio sostituendo gli inibitori della calcineurina con gli inibitori del pathway di PI3K-AKT-mTOR). Tuttavia, diminuire l’immunodepressione negli individui con KS iatrogeno può aumentare il rischio di rigetto del trapianto. Molti studi hanno riportato che nel KS iatrogeno il trattamento con rapamicina (un inibitore di mTOR) porta alla scomparsa delle lesioni, dal momento che tra i pathway attivati dalla proteina vGPCR è incluso anche PI3K-AKT-mTOR.
I trattamenti diretti ai tumori sono necessari nei pazienti con KS associato all’AIDS e con KS iatrogeno in cui gli agenti che bersagliano il sistema immunitario sono insufficienti, e nei pazienti con KS classico ed endemico. L’evidenza di alta qualità per la gestione clinica del KS riguarda solo il management del KS associato all’AIDS; l’approccio clinico per il trattamento dei pazienti con altre forme di KS si fonda prevalentemente su piccole descrizioni retrospettive di casi e sull’esperienza dei clinici piuttosto che su dati sperimentali.
Negli individui HIV positivi la terapia HAART abbassa la carica virale, consente un incremento dei linfociti T CD4 e CD8, una normalizzazione dei linfociti B, portando quindi a una ricostituzione del sistema immunitario. Questi effetti sono connessi a una significativa diminuzione dell’incidenza dei tumori associati all’AIDS, in cui è incluso il KS. Gli esiti del regime terapeutico HAART nell’incidenza e nella regressione dei tumori sono stati assiduamente studiati in gruppi di pazienti trattati con inibitori della trascrittasi inversa e con inibitori delle proteasi. Per quanto concerne il KS, si è visto che entrambe le classi di farmaci riducono le lesioni del KS ma la remissione completa avviene più frequentemente nei pazienti trattati con inibitori delle proteasi rispetto a quelli trattati con inibitori della trascrittasi inversa. Questi ultimi avevano un basso numero di linfociti T CD4 all’inizio dello studio, e dopo la terapia si vedeva solo un piccolo e parziale rialzo dei CD4; quindi nei pazienti con una bassa attività immunitaria, gli inibitori delle proteasi erano più efficienti nell’indurre una risposta completa. Sono stati fatti molti studi il cui scopo era quello di identificare le azioni non virologiche degli inibitori delle proteasi, tra cui ritonavir, squinavir, indinavir e nelfinavir. Si è scoperto che questi farmaci colpiscono direttamente svariati fattori coinvolti nella proliferazione, nell’angiogenesi, nella sopravvivenza, nell’invasione e nell’infiammazione. Per esempio, per contrastare l’angiogenesi e l’invasività, i bersagli sono il fattore di crescita di base dei fibroblasti (bFGF), il VEGF e le metalloproteasi della matrice (MMP). Per limitare l’infiammazione, invece, i target sono la selettina E, le molecole di adesione ICAM e VCAM, e diverse citochine (TNFα, IL-6 e IL-8). Oltre a ciò, gli inibitori delle proteasi sono in grado di bloccare il proteasoma, un complesso multiproteico che regola diversi pathway, tra cui il turnover proteico, l’eliminazione delle proteine mal ripiegate, l’apoptosi, la degradazione dei prodotti dei geni oncosoppressori, la funzione degli inibitori delle chinasi ciclina-dipendente (CDK). Il blocco del proteasoma può arrestare la proliferazione e la sopravvivenza delle cellule tumorali, e può rendere tali cellule più sensibili alle radiazioni ionizzanti. Gli studi hanno rivelato che gli inibitori delle proteasi agiscono come inibitori reversibili del proteasoma, come altri potenti farmaci utilizzati per la terapia dei tumori (ad esempio il bortezomib).
Per il KS associato all’AIDS sono state approvate due terapie patogenesi-dirette, ovvero l’interferone alfa (IFNα) e l’alitretinoina (un agonista del recettore dell’acido retinoico). Il primo è una citochina dagli effetti antiproliferativi e antivirali che possono diminuire l’angiogenesi e modulare le risposte immunitarie umorali e cellulari dell’ospite. Oggi si usa raramente, in quanto ci sono degli agenti alternativi che si somministrano con più facilità e hanno profili più favorevoli di effetti indesiderati. Il secondo, che si applica mediante gel, induce l’apoptosi delle cellule di KS in vitro, e si pensa che inibisca la proliferazione cellulare e promuova il differenziamento.
Fonti
- https://thedarkpark.wordpress.com/2011/09/07/kaposi%E2%80%99s-sarcoma-associated-herpesvirus/
- Patrick S. Moore and Yuan Chang. 1995 “Detection of herpesvirus-like DNA sequences in Kaposi’s sarcoma in patients with and those without hiv infection”, The New England Journal of Medicine
- Ethel Cesarman, Blossom Damania, Susan E. Krown, Jeffrey Martin, Mark Bower and Denise Whitby. 2019. “Kaposi Sarcoma”, Nature
- William Harrington, Laura Sieczkowski, Carlos Sosa, Sharon Chan-a-Sue, Jiang P. Cai, Lisa Cabral, Charles Wood. 1997. “Activation of HHV-8 by HIV-1 tat”, Lancet
- Silvia Montaner, Akrit Sodhi, Alfredo Molinolo, Thomas H. Bugge, Earl T. Sawai, Yunsheng He, Yi Li, Patricio E. Ray, and J. Silvio Gutkind. 2003. “Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi’s sarcomagenesis and can promote the tumorigenic potential of viral latent genes”, Cencer Cell
- Silvia Montaner, Akrit Sodhi, Amanda K. Ramsdell, Daniel Martin, Jiadi Hu, Earl T. Sawai, and J. Silvio Gutkind. 2006. “The Kaposi’s Sarcoma–Associated Herpesvirus G Protein–Coupled Receptor as a Therapeutic Target for the Treatment of Kaposi’s Sarcoma”, Cancer Research
- Patrick S. Moore and Yuan Chang. 2003. “Kaposi’s sarcoma–associated herpes virus immunoevasion and tumorigenesis: two sides of the same coin?”, Annu Rev Microbiol
- Paul E. Wakely, Geetha Menezes and Gerard J. Nuovo. 2002. “Primary Effusion Lymphoma: Cytopathologic Diagnosis Using In Situ Molecular Genetic Analysis for Human Herpesvirus 8”, Modern Pathology
- https://askhematologist.com.com/castleman-disease/
- Paolo Monini, Cecilia Sgadari, Elena Toschi, Giovanni Barillari and Barbara Ensoli. 2004. “Antitumour effects of antiretroviral therapy”, Nature
Sono affetto da Herpes virus tipo 8 da due anni. Sono in cura oncologica. Vorrei sapere di più su malattia Castleman. Grazie
Buonasera Leonardo. Chiedo scusa se rispondo tardi ma ho visto solo ora il suo commento. Riguardo ai disordini di Castleman, non c’è molto da dire rispetto a ciò che ho scritto. Comunque, non si tratta di veri e propri tumori, in quanto non c’è una proliferazione di un singolo clone di cellule, bensì si verifica un’espansione policlonale. Motivo per cui si chiamano disturbi linfoproliferativi. Potrebbero incrementare il rischio di insorgenza di tumori come i linfomi
Olá. Peço por gentileza que me envie este artigo completo por email. Sou mestrando e pesquisador na área da imunogenética. Estou desenvolvendo um estudo sobre dois tipos de herpesvírus, 6 e 8.